Pathological Mechanisms of Alzheimer's Disease (AD)
Release time:2024-10-15 17:10:51
Alzheimer's disease (AD) is the most common neurodegenerative disorder and one of the leading types of dementia. Since Dr. Alois Alzheimer first described the disease in 1901, AD has posed a significant challenge to the medical field, with no cure found thus far. To conquer any disease, understanding its pathological mechanisms is crucial for developing the most effective treatment strategies.
Below, we delve into the pathological mechanisms of AD based on a review article by Dr. Wang Xin and Dr. Zheng Qiuyang from Xiamen University, published in Protein & Cell, titled Alzheimer's Disease: Insights into Pathology, Molecular Mechanisms, and Therapy. This review offers insights into the complex mechanisms underlying AD and provides a foundation for potential therapeutic approaches aimed at addressing this major medical challenge.
The main pathological features of AD include amyloid plaques formed by β-amyloid (Aβ) and neurofibrillary tangles (NFTs) caused by hyperphosphorylated tau protein. Additionally, AD is characterized by synapse and neuron loss, along with reactive gliosis.
1. Amyloid β
Aβ is derived from its precursor protein, APP, through sequential cleavage by β- and γ-secretases. Due to the hydrophobic nature of its amino acids, Aβ monomers tend to aggregate into oligomers, eventually forming fibrils and amyloid plaques. Recently, cryo-electron microscopy revealed two distinct types of Aβ42 fibrils in the human brain: Type I fibrils are primarily found in sporadic AD patients, while Type II fibrils are mostly present in familial AD cases. Aβ initiates the fibrillation process in a prion-like manner, forming misfolded β-sheet structured Aβ seeds, which act as templates for larger amyloid aggregates. In 1992, Hardy and Higgins proposed the Aβ cascade hypothesis, suggesting that excessive production and aggregation of Aβ in the brain is the initial trigger for AD, leading to the formation of NFTs, neuronal loss, and ultimately cognitive decline. While the Aβ hypothesis has been questioned, strong genetic evidence supports Aβ's central role in the disease. Recently, clinical trials of monoclonal antibodies targeting Aβ have been successful 【2-4】, providing crucial clinical support for the Aβ cascade hypothesis.
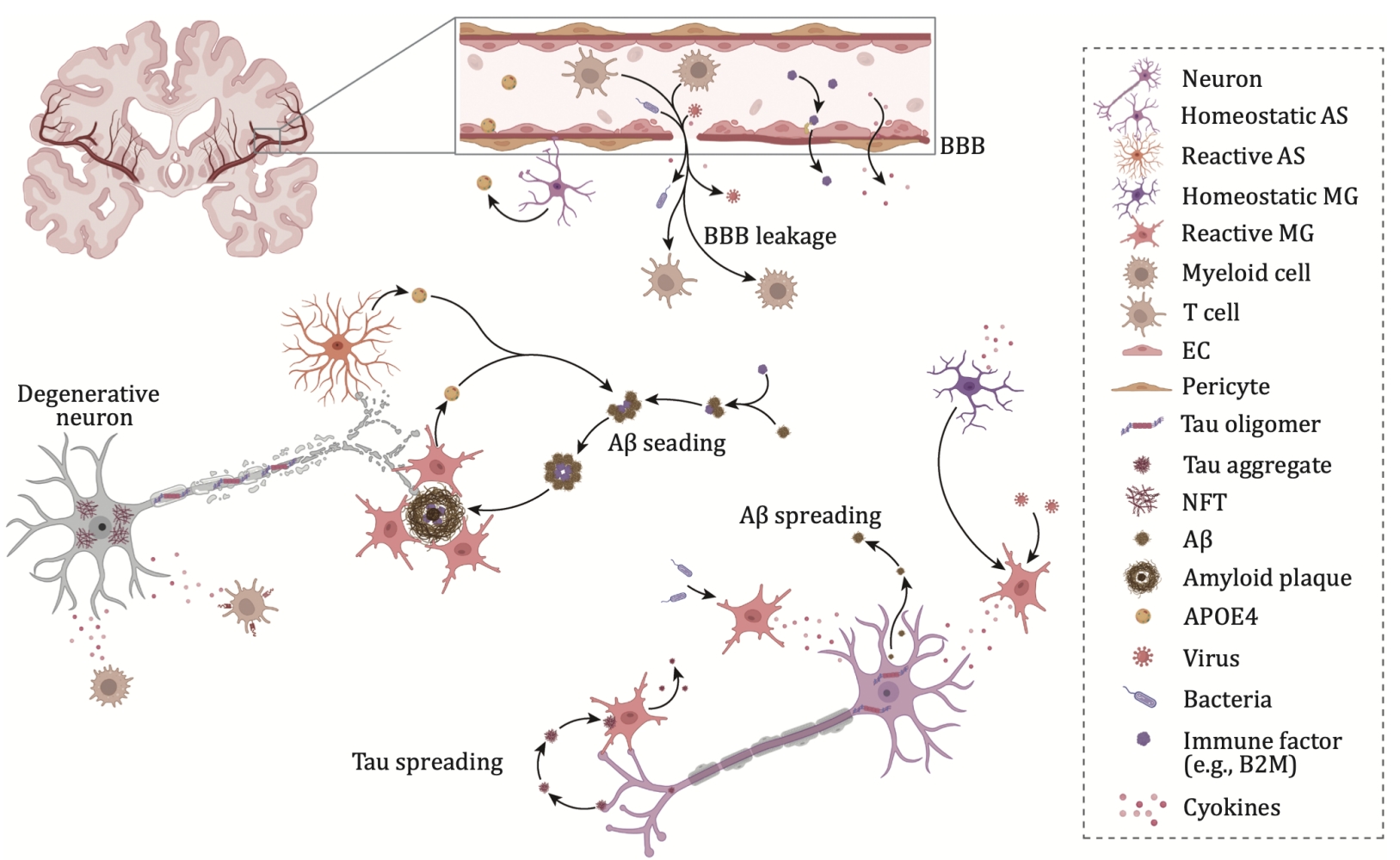
For a long time, Aβ has been regarded as the main pathogenic factor in amyloid plaques. However, recent research shows that β2-microglobulin (B2M) is another amyloid protein with a structure similar to Aβ. B2M is not only produced in the brain but can also come from peripheral blood and cross the blood-brain barrier. The B2M-Aβ co-aggregates, formed by the interaction of B2M and Aβ, exhibit stronger neurotoxicity. Studies have shown that knocking out the B2M gene significantly reduces Aβ neurotoxicity. Moreover, B2M is highly expressed in peripheral immune cells and can be released into the bloodstream, crossing the blood-brain barrier into the brain. Clearing peripheral B2M has been shown to effectively improve cognitive function in AD mouse models【5,6】. These findings offer a new perspective on amyloid pathology and provide fresh insights and therapeutic possibilities for Alzheimer's disease research.
2. Abnormal Modifications of Tau Protein
Tau protein, the primary component of NFTs, normally functions as a microtubule-associated protein that plays a key role in regulating microtubule stability and axonal transport. The progression of brain atrophy in AD is closely correlated with the accumulation of tau, even more so than with Aβ deposition. NFTs consist of paired helical filaments (PHFs) and straight filaments (SFs), with notable differences in the arrangement of these fibrils. Various tauopathies, including AD, chronic traumatic encephalopathy (CTE), corticobasal degeneration (CBD), Pick's disease (PiD), and progressive supranuclear palsy (PSP), exhibit different folding patterns of tau, highlighting the diversity of tau pathology. The spread of tau from cell to cell involves the self-replication of primary tau seeds, which induce the accumulation and templated fibrillation of endogenous tau, allowing abnormal tau to propagate through the brain in a prion-like manner. In AD, NFT pathology begins in the entorhinal cortex and perirhinal cortex and follows a predictable pattern of spreading to the hippocampus and neocortex. Tau aggregation is influenced by interactions among its post-translational modifications (PTMs), genetic mutations, and aggregation inducers, leading to the formation of toxic NFTs within neurons. In addition to the common phosphorylation modification of tau, recent studies have identified a ubiquitin-like modification known as UFMylation as playing a key role in tau pathology. Reducing the expression of UFMylation-related genes can decrease the formation of tau inclusions in iPSC-derived neurons carrying tau mutations and reduce tau spread in the brains of PS19 mice【7,8】. This suggests that targeting UFMylation could be a potential therapeutic strategy for treating tau-related diseases.
3.Neuronal Loss
Extensive neuronal loss, especially in brain regions critical for memory and higher cognitive functions, is a hallmark pathological feature of AD. In the AD brain, various forms of regulated cell death (RCD) pathways, including necroptosis, pyroptosis, apoptosis, ferroptosis, and autophagy-dependent cell death, are abnormally activated. Recent research has found that the neuron-specific long non-coding RNA MEG3 is upregulated in AD patients, leading to neuronal necroptosis. Downregulating MEG3 can inhibit neuronal death in the brains of AD mouse models transplanted with human neurons【9】. Additionally, Aβ can induce neuron re-entry into the cell cycle in a tau-dependent manner, causing synaptic dysfunction and neuronal death. Cell cycle re-entry is an early critical event leading to neuronal loss in AD.
4. Demyelination
Myelin is a spiral membrane structure tightly wrapped around axons, formed by oligodendrocytes. In the preclinical stage of AD, early signs of myelin damage appear in the cerebral cortex, suggesting that myelin injury could be an early indicator of brain pathology. Moreover, a specific subset of disease-associated oligodendrocytes (DAOs) has been identified in AD mouse models and patients, playing a key role in disease pathology. Myelin dysfunction and demyelination can accelerate amyloid plaque deposition in AD mouse models treated with EAE and Cuprizone【10】, indicating the potential of preserving oligodendrocyte health and myelin integrity as a therapeutic strategy to slow AD progression.
Figure 1. Impact of systemic inflammation on AD pathogenesis.
5. Reactive Gliosis and Neuroinflammation
Increasing evidence suggests that reactive astrogliosis and microglial proliferation are significant pathological features of Alzheimer's disease (AD) and play a key role in its pathogenesis. The APOE ε4 allele is the most important genetic risk factor for sporadic AD, increasing the risk of developing the disease by 3 to 15 times. This gene is primarily expressed by astrocytes and microglia in the central nervous system. Additionally, several single nucleotide polymorphisms (SNPs) and rare coding variants related to the immune system, which are thought to affect microglial function, have been identified as AD risk factors. These include TREM2, BIN1, CLU, CR1, PICALM, CD33, and the MS4A gene cluster.
Microglia are the brain’s resident immune cells. In the AD brain, reactive microglia gather around amyloid plaques; tau pathology is also closely associated with microglial activation. A subset of disease-associated microglia (DAM) has been identified in AD mouse models, located near amyloid plaques, and may limit neurodegeneration. In AD patients carrying the APOE ε4/ε4 genotype, a lipid-droplet-accumulating microglial subset (LDAM) has been identified. Additionally, terminal inflammatory microglia (TIM) and degenerative (senescent) microglial subsets related to Aβ clearance and tau pathology have been discovered, highlighting the diverse roles of microglia in AD.
Astrocytes play key roles in maintaining extracellular fluid and neurotransmitter homeostasis, inducing synapse formation, and providing neurotrophic support. A disease-associated astrocyte (DAA) subset has been identified in AD mouse models and the aged human brain. Furthermore, tau oligomers can induce astrocytes to release HMGB1, leading to astrocyte senescence. This suggests that a decline in astrocytic surveillance functions may contribute to accelerated aging in the progression of Alzheimer's disease.
The interpretation of Alzheimer's disease (AD) pathological mechanisms above is sourced from the Brainnews media team. The Brain Case team has been engaged in viral vector research and large-scale production for many years. By using viral vectors to deliver foreign genes into animals, they aim to model and treat AD. For more details, please contact BD@ebraincase.com
Ref:
[1] Zheng, Q. & Wang, X. Alzheimer's disease: insights into pathology, molecular mechanisms, and therapy. Protein Cell (2024). https://doi.org/10.1093/procel/pwae026 [2] Budd Haeberlein, S. et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer's Disease. J Prev Alzheimers Dis 9, 197-210 (2022). https://doi.org/10.14283/jpad.2022.30 [3] van Dyck, C. H. et al. Lecanemab in Early Alzheimer's Disease. N Engl J Med 388, 9-21 (2023). https://doi.org/10.1056/NEJMoa2212948 [4] Sims, J. R. et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 330, 512-527 (2023). https://doi.org/10.1001/jama.2023.13239 [5] Zhao, Y. et al. beta(2)-Microglobulin coaggregates with Abeta and contributes to amyloid pathology and cognitive deficits in Alzheimer's disease model mice. Nat. Neurosci. 26, 1170-1184 (2023). https://doi.org/10.1038/s41593-023-01352-1 [6] Gao, Y. et al. beta2-microglobulin functions as an endogenous NMDAR antagonist to impair synaptic function. Cell 186, 1026-1038 e1020 (2023). https://doi.org/10.1016/j.cell.2023.01.021 [7] Parra Bravo, C. et al. Human iPSC 4R tauopathy model uncovers modifiers of tau propagation. Cell (2024). https://doi.org/10.1016/j.cell.2024.03.015 [8] Samelson, A. J. et al. CRISPR screens in iPSC-derived neurons reveal principles of tau proteostasis. bioRxiv (2023). https://doi.org/10.1101/2023.06.16.545386 [9] Balusu, S. et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer's disease. Science 381, 1176-1182 (2023). https://doi.org/10.1126/science.abp9556 [10] Depp, C. et al. Myelin dysfunction drives amyloid-beta deposition in models of Alzheimer's disease. Nature 618, 349-357 (2023). https://doi.org/10.1038/s41586-023-06120-6