1. Social Defeat Stress Induces Mitochondrial Dysfunction in the mPFC of Stress-Susceptible Adolescent Mice
Wild-type C57BL/6J mice aged 35–44 days were subjected to an accelerated social defeat stress (AcSD) paradigm followed by a social interaction test (SIT). Based on behavioral outcomes, the mice were categorized into susceptible, resilient, and control groups (Fig. 1a–e). Compared with control and resilient mice, the susceptible group exhibited significantly lower sucrose preference (Fig. 1f–g), indicating anhedonia.
To assess mitochondrial function, researchers measured mitochondrial membrane potential (MMP) and ATP levels in the medial prefrontal cortex (mPFC) of mice that underwent AcSD or control treatment. Results showed that both male and female susceptible mice exhibited significantly reduced MMP and ATP levels in the mPFC, with no significant changes observed in the hippocampus (Fig. 1h–j). These findings suggest that social stress induces mitochondrial dysfunction in the mPFC of adolescent mice displaying depression-like phenotypes, with no sex-specific differences observed.
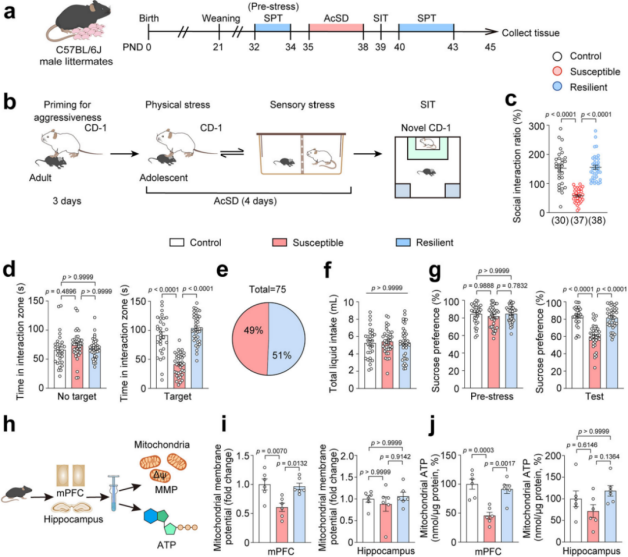 Figure 1. Stress Induces Mitochondrial Dysfunction in the mPFC of Adolescent Mice with Depression-like Phenotypes
Figure 1. Stress Induces Mitochondrial Dysfunction in the mPFC of Adolescent Mice with Depression-like Phenotypes
To determine which aspects of mitochondrial dysfunction contribute to adolescent depression, researchers administered the mitochondrial fission inhibitor Mdivi-1 and the PPAR-δ agonist GW0742 to stress-susceptible mice. The results showed that Mdivi-1 significantly alleviated social avoidance and anhedonia-like behaviors induced by AcSD, while GW0742 had no such effect (Fig. 2k–n).
Mdivi-1 is known to inhibit mitochondrial fission by blocking the self-assembly of DRP1 (Dynamin-related protein 1, gene name: Dnm1l). Further analysis revealed that Mdivi-1 restored mitochondrial function, improved mitochondrial morphology, and suppressed DRP1 expression in the mPFC of susceptible adolescent mice following AcSD, whereas GW0742 did not show these effects.
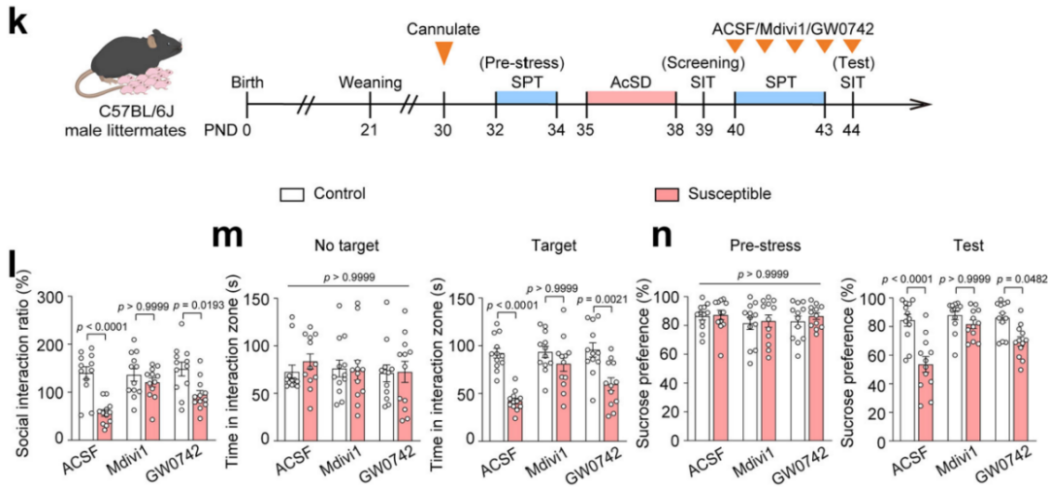 Figure 2. Mdivi-1 Restores Mitochondrial Function Impaired by AcSD
Figure 2. Mdivi-1 Restores Mitochondrial Function Impaired by AcSD
2. DRP1 in mPFC Neurons Contributes to Stress Susceptibility in Adolescent Mice
Cellular Localization Experiments:
Researchers further examined the expression levels of DRP1 protein and Dnm1l mRNA in AcSD-exposed mice. They found that both phosphorylated and total DRP1 protein levels, as well as Dnm1l mRNA expression, were significantly elevated in the mPFC of stress-susceptible mice. Moreover, Dnm1l mRNA levels were negatively correlated with social interaction ratios (Fig. 3a–c).
During AcSD exposure of varying durations, DRP1 expression increased over time. A 4-day AcSD protocol led to reduced social interaction, which could be reversed by the antidepressant fluoxetine. These findings suggest that DRP1 is closely associated with depression-like behaviors during adolescence and plays a critical role in mediating mitochondrial dysfunction in the mPFC, thereby facilitating stress-induced behavioral changes.
Analysis of human brain samples revealed elevated Dnm1l expression in the dorsolateral prefrontal cortex of individuals with depression.
In brain slices from AcSD mice, immunofluorescence co-labeling of DRP1 with NeuN (neuronal marker), Iba1 (microglia), and GFAP (astrocytes) revealed that DRP1 was highly colocalized with NeuN+ neurons in the mPFC of stress-susceptible adolescent mice (Fig. 3d–i). Importantly, DRP1 intensity in NeuN+ cells was negatively correlated with social interaction (Fig. 2e), highlighting a direct link between neuronal DRP1 in the mPFC and depression-like behavior in adolescent mice.
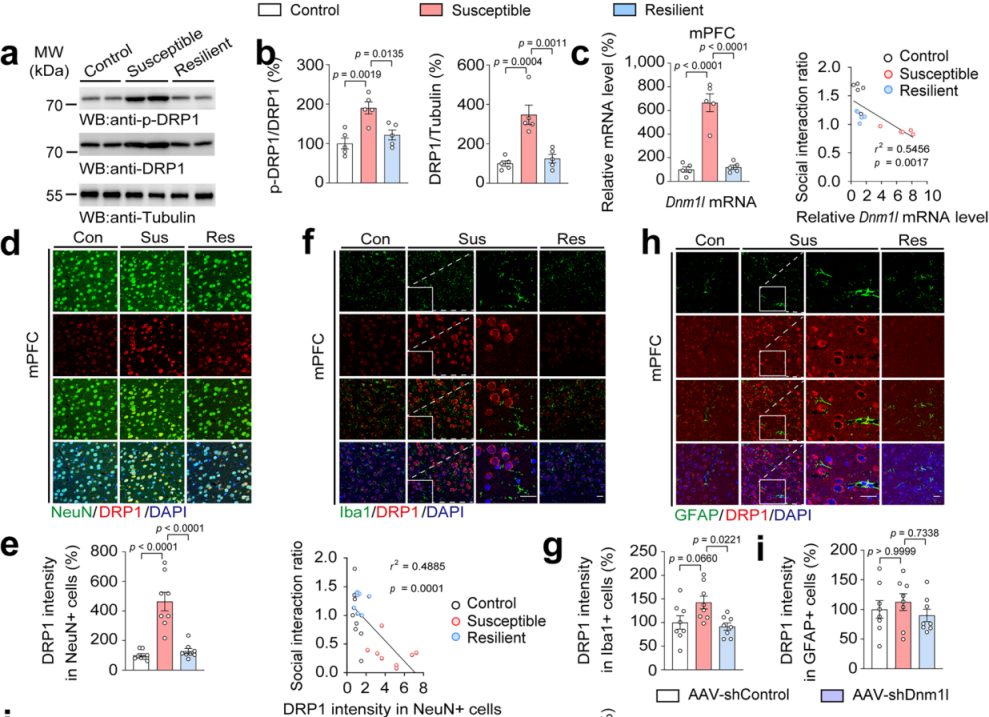 Figure 3. DRP1 Protein and mRNA Levels Are Elevated in mPFC Neurons of Stress-Susceptible Mice
Figure 3. DRP1 Protein and mRNA Levels Are Elevated in mPFC Neurons of Stress-Susceptible Mice
DRP1 Knockdown and Overexpression Experiments:
On postnatal day 29 (PND29), AAV-hSyn-shDnm1l-EGFP or control virus AAV-hSyn-shControl-EGFP was injected into the mPFC of wild-type mice to knock down DRP1 specifically in neurons (Fig. 4j). Knockdown of DRP1 restored AcSD-impaired mitochondrial function and significantly improved social interaction ratios and sucrose preference (Fig. 4k–n). This indicates that DRP1 knockdown alleviates mitochondrial dysfunction and depression-like behaviors caused by AcSD.
Conversely, AAV-hSyn-Dnm1l-EGFP or control AAV-hSyn-EGFP was used to overexpress DRP1 specifically in mPFC neurons under subthreshold social defeat stress (SSD) conditions (Fig. 4o). Overexpression of DRP1 led to mitochondrial dysfunction in the mPFC, and significantly reduced social interaction and sucrose preference (Fig. 4p–s). These results demonstrate that increased DRP1 expression in mPFC neurons enhances stress susceptibility in adolescent mice.
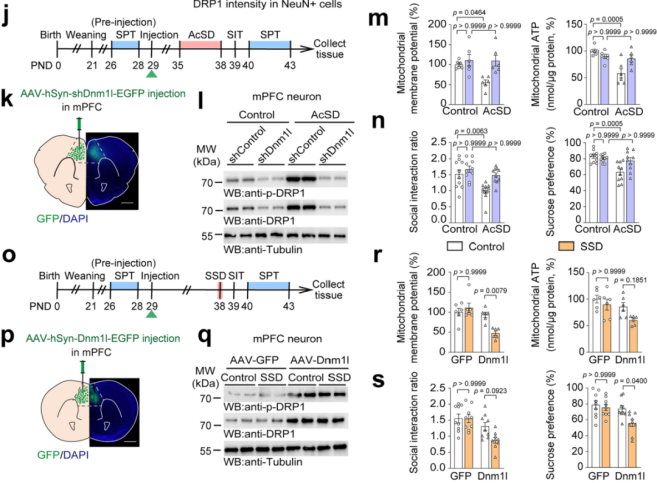 Figure 4. DRP1 in mPFC Neurons Regulates Stress Susceptibility in Adolescent Mice
Figure 4. DRP1 in mPFC Neurons Regulates Stress Susceptibility in Adolescent Mice
3. Neuronal C/EBPβ Acts as a Transcriptional Activator of Dnm1l and Regulates Stress Susceptibility in Adolescent Mice
Transcription Factor Screening:
Based on the ChIP-Atlas database, researchers screened potential transcription factors that bind to the Dnm1l promoter in HeLa cells. Candidates included Gabpa, Ctcf, Rest, and Cebpb (Fig. 5a). Among them, only Cebpb showed significantly increased mRNA levels in stress-susceptible mice compared to controls (Fig. 5b), and its expression was negatively correlated with the social interaction ratio (Fig. 5c). Protein analysis further revealed increased total and phosphorylated C/EBPβ levels in the mPFC of susceptible mice (Fig. 5d).
Fluorescence in situ hybridization (FISH) combined with immunostaining showed elevated levels of both p-C/EBPβ and Dnm1l mRNA in the mPFC of susceptible mice, and p-C/EBPβ intensity was negatively correlated with social interaction (Fig. 5e–f), suggesting a strong correlation between p-C/EBPβ and Dnm1l mRNA (Fig. 5e–g). Immunostaining also showed a positive correlation between C/EBPβ and DRP1 protein levels (Fig. 5h–j), indicating that C/EBPβ may act as a transcriptional regulator of DRP1 in the mPFC of stress-susceptible mice.
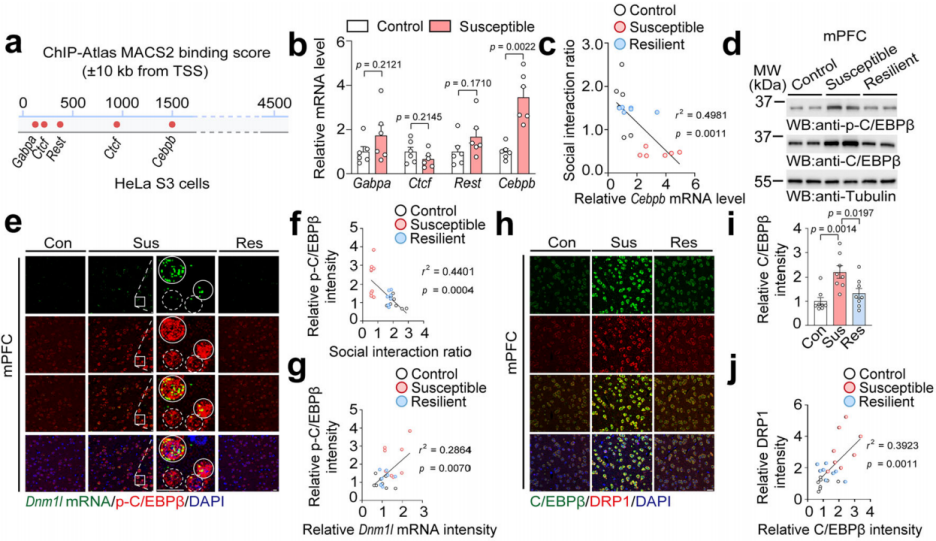
Fig. 5 C/EBPβ levels are significantly elevated in susceptible mice and positively correlated with DRP1 expression.
Binding Site and Activity Validation:
ChIP-seq analysis revealed high binding scores for C/EBPβ at the Dnm1l promoter (Fig. 6k). Researchers constructed various truncated Dnm1l promoters and conducted luciferase reporter assays to identify the core binding region (-1369 to -1060) (Fig. 6l–o). Electrophoretic mobility shift assays (EMSA) and chromatin immunoprecipitation (ChIP) confirmed that C/EBPβ specifically binds to the Dnm1l promoter (Fig. 6p–q).
C/EBPβ has three isoforms: LAP*, LAP (activators), and LIP (inhibitor). Using C/EBPβ LAP transgenic (Tg) mice (which overexpress LAP isoform under the Thy1 promoter to mimic neuronal overactivation of C/EBPβ) and primary mouse cortical neurons, the researchers found that C/EBPβ knockout decreased DRP1 mRNA and protein levels, while LAP overexpression increased them, confirming that C/EBPβ promotes DRP1 expression as a transcriptional activator of Dnm1l (Fig. 6r–w).
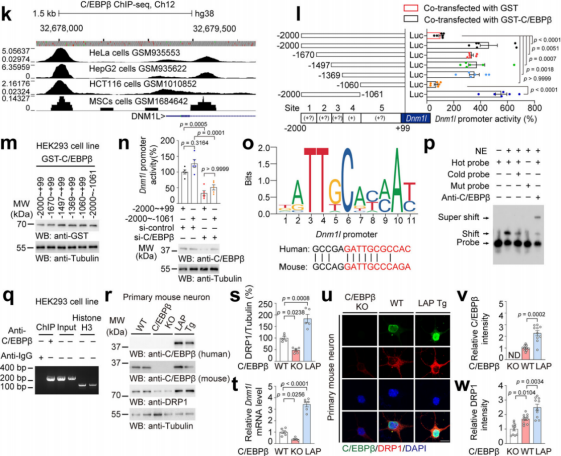 Fig. 6 C/EBPβ promotes DRP1 expression by acting as a transcriptional activator of Dnm1l.
Fig. 6 C/EBPβ promotes DRP1 expression by acting as a transcriptional activator of Dnm1l.
Cellular Localization and Correlation Analysis:
Immunostaining showed that C/EBPβ was most highly expressed in NeuN⁺ neurons in the mPFC of susceptible mice (Fig. 7a–b), and its intensity in these cells was negatively correlated with the social interaction ratio (Fig. 7c), suggesting that C/EBPβ functions primarily in neurons as a transcription factor for DRP1.
To explore the behavioral impact of neuronal C/EBPβ, wild-type (WT; C/EBPβ⁺/⁺), C/EBPβ heterozygous knockout (C/EBPβ⁺/⁻), and C/EBPβ LAP transgenic (LAP Tg) mice were used. AAV-hSyn-EGFP or AAV-hSyn-Dnm1l-EGFP was injected into the mPFC of WT and C/EBPβ⁺/⁻ mice, followed by AcSD exposure, behavioral testing, and mitochondrial analysis (Fig. 7d–g).
Results showed that DRP1 protein levels in mPFC neurons were significantly reduced in C/EBPβ⁺/⁻ mice (Fig. 7f–g), and these mice exhibited improved mitochondrial membrane potential and ATP levels after AcSD. However, DRP1 overexpression reversed these improvements, indicating that C/EBPβ deletion alleviates mitochondrial dysfunction by downregulating DRP1 (Fig. 7h–i).
Moreover, C/EBPβ⁺/⁻ mice showed increased social interaction and sucrose preference, which disappeared when DRP1 was overexpressed. This demonstrates that lowering C/EBPβ in the mPFC reduces adolescent stress susceptibility (Fig. 7j–k).
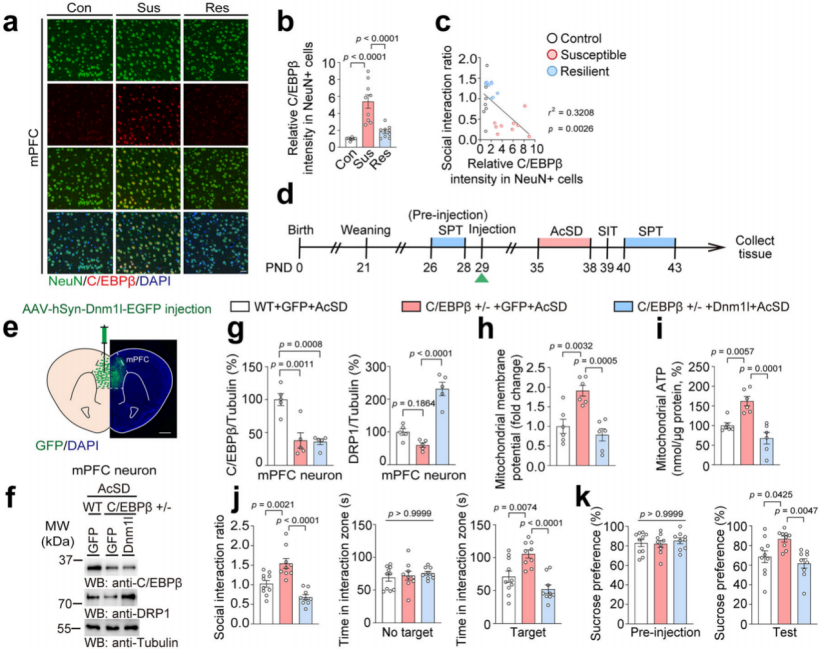
Fig. 7 Reducing C/EBPβ in the mPFC lowers stress susceptibility in adolescent mice.
Next, AAV-hSyn-shDnm1l-EGFP or AAV-hSyn-shControl-EGFP was injected into the mPFC of LAP Tg and WT mice, followed by SSD or control treatments (Fig. 8m). After SSD, DRP1 levels increased in LAP Tg mice but not in WT mice (Fig. 8l, n–o). Knocking down DRP1 in LAP Tg neurons alleviated SSD-induced mitochondrial dysfunction, indicating that neuronal C/EBPβ aggravates mitochondrial dysfunction via DRP1 upregulation (Fig. 8p–q).
Likewise, LAP Tg mice exposed to SSD exhibited reduced social interaction and sucrose preference, suggesting that neuronal C/EBPβ overexpression enhances vulnerability to depression-like behavior in adolescence (Fig. 8r–s).
Together, these findings demonstrate that C/EBPβ promotes DRP1 expression in mPFC neurons and contributes to adolescent depression-like behavior.
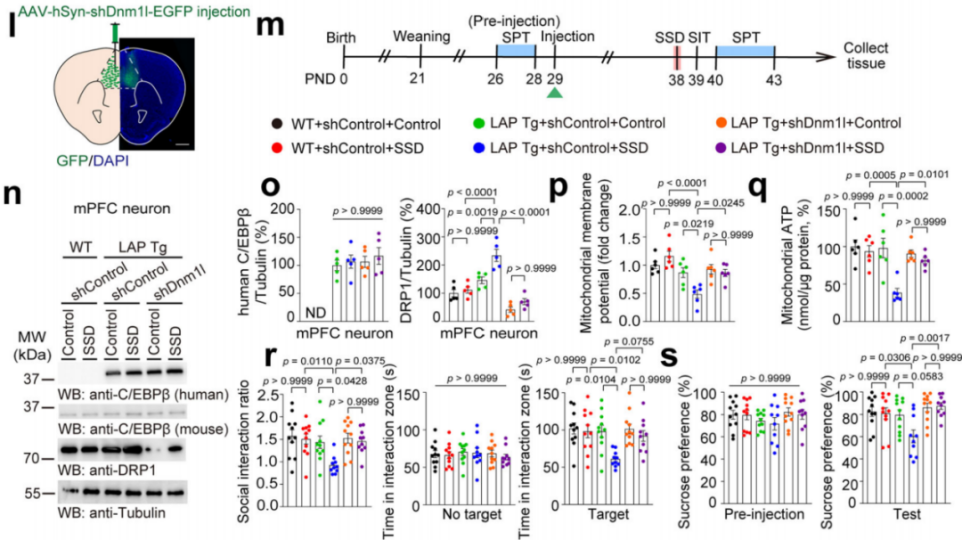 Fig. 8 C/EBPβ promotes adolescent depression-like behavior by enhancing DRP1 expression.
Fig. 8 C/EBPβ promotes adolescent depression-like behavior by enhancing DRP1 expression.
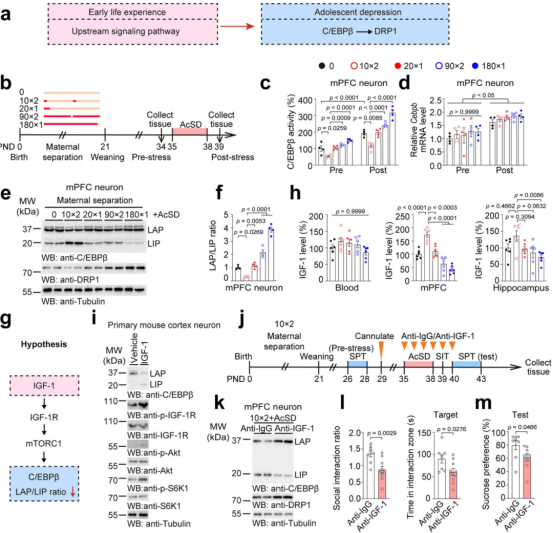
4. Different Maternal Separation Paradigms Trigger Distinct Stress Susceptibility Outcomes via IGF-1–Mediated C/EBPβ Activity
The previous findings showed that overactivation of C/EBPβ increases stress susceptibility in adolescent mice, but its upstream mechanisms remained unclear. Epidemiological and preclinical studies suggest that early life stressors influence adolescent stress resilience, and the “two-hit” model provides a conceptual framework for exploration (Fig. 9a).
Maternal separation (MS), a classical early life stress model, can modulate stress susceptibility during adolescence. Therefore, the researchers shifted from the AcSD model to the MS model to investigate the upstream signaling that promotes neuronal C/EBPβ activity.
They found that MS altered C/EBPβ activity and the LAP/LIP ratio in mPFC neurons of adolescent mice (Fig. 9b–f). Even in stress-susceptible mice without early-life stress, this ratio was increased, suggesting its involvement in C/EBPβ activation and in the influence of early life experiences on adolescent depression.
Additionally, IGF-1 may regulate the LAP/LIP ratio of C/EBPβ through the IGF-1R/mTORC1 signaling pathway (Fig. 9g). Low IGF-1 levels were observed in both MS-exposed and non-MS stress-susceptible mice, indicating that IGF-1 links these two types of stressors across different developmental timelines.
Treatment of primary mouse cortical neurons with IGF-1 revealed that IGF-1 increased the phosphorylation-to-total protein ratio of IGF-1 receptor (IGF-1R) and other related proteins (Fig. 9h–i). This confirms that IGF-1 binds to its receptor and activates the mTORC1 pathway, which in turn reduces the LAP/LIP ratio and downregulates C/EBPβ activity, without changing total C/EBPβ protein or mRNA levels. This reduction was associated with a decline in Dnm1l mRNA expression (Fig. 9i).
To investigate IGF-1’s role in stress susceptibility, adolescent mice were subjected to both MS and AcSD, and intracerebrally injected with anti–IGF-1 or anti-IgG to block brain IGF-1 (Fig. 9j). Blocking IGF-1 restored neuronal LAP/LIP ratios and DRP1 mRNA/protein levels, and reduced social interaction ratios and other stress-induced behavioral deficits (Fig. 9k–m).
Further experiments combining anti–IGF-1 antibody and AcSD revealed a synergistic worsening of these changes. However, 10min×2 MS (a mild maternal separation paradigm) suppressed the C/EBPβ–DRP1 axis and rescued behavioral impairments in adolescent mice exposed to anti–IGF-1, AcSD, or both. This suggests that IGF-1 upregulation induced by 10min×2 MS is crucial for maintaining stress resilience during adolescence.
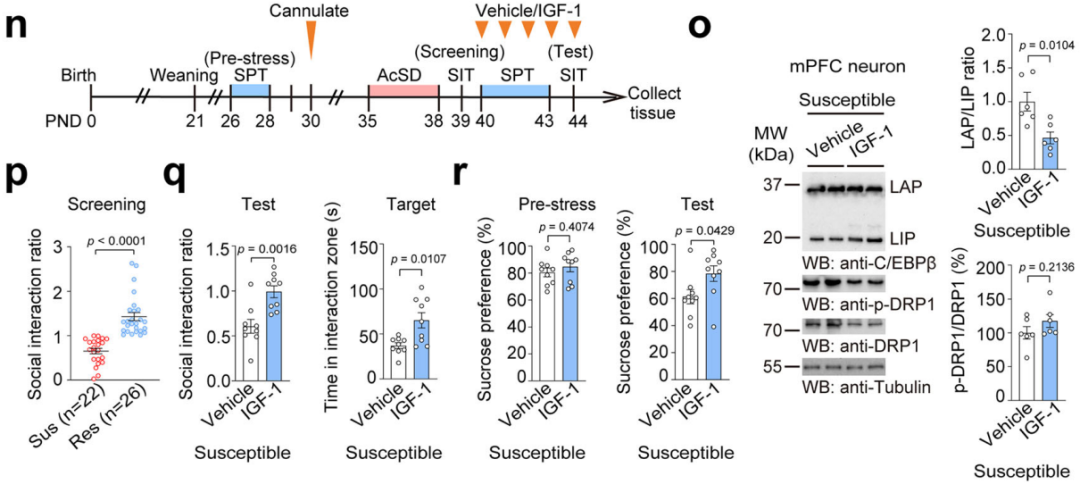
5. Maternal Behavior Prevents Stress Susceptibility in Adolescent Mice by Upregulating mPFC IGF-1 Levels
Maternal separation can alter maternal behavior. After quantifying maternal behavior, the researchers found that the 10min×2 maternal separation group had an increased number of maternal behavior samples, while the 180min×1 maternal separation group had a decreased number (Fig. 11a). Furthermore, the number of maternal behavior samples was positively correlated with the social interaction ratio and IGF-1 levels in the mPFC (Fig. 11b–d), indicating that maternal behavior is crucial for regulating IGF-1 levels in the mPFC and protecting mice from stress susceptibility.
Using the non-lactating mother (LnL) model, which increases maternal behavior towards offspring, the researchers conducted 180min×1 maternal separation and AcSD paradigms (Fig. 11e). The results showed that LnL mothers exhibited more maternal behavior (Fig. 11f), leading to higher IGF-1 levels in the mPFC (Fig. 11g), lower LAP/LIP ratios, reduced DRP1 protein expression, and lower Dnm1l mRNA levels (Fig. 11h). This improved the social avoidance and sucrose preference deficits induced by prolonged maternal separation (Fig. 11i–j), with no impact on offspring weight. This suggests that increased maternal care can suppress the C/EBPβ–DRP1 axis.
Further experiments investigating the role of IGF-1 in maternal behavior’s anti-depressant effects confirmed that maternal behavior elevates IGF-1 levels, which, in turn, regulates the LAP/LIP ratio to prevent stress susceptibility.
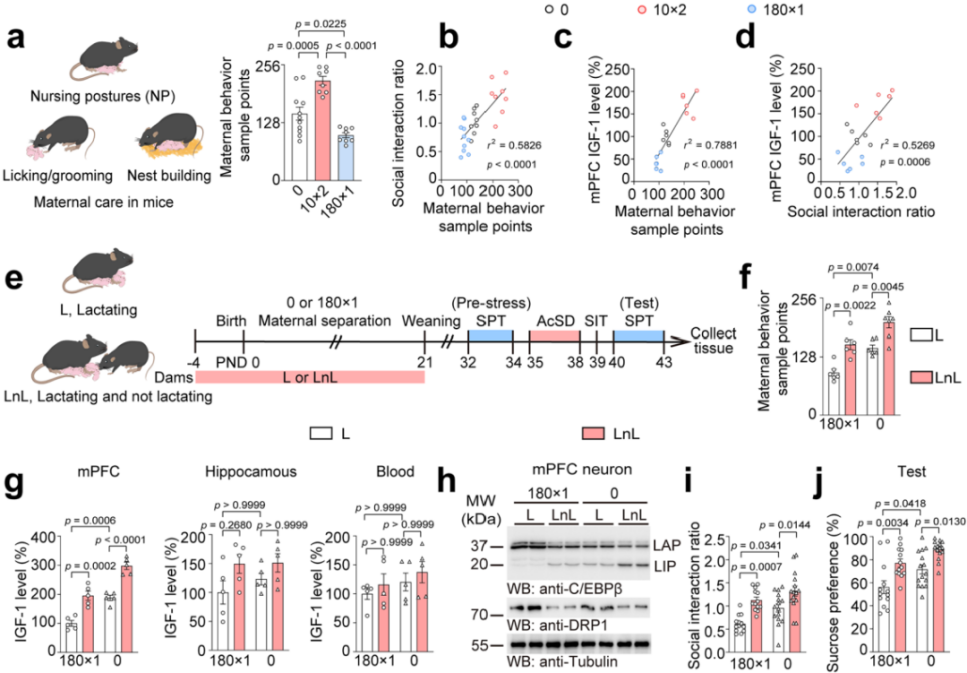 Fig. 11 Maternal behavior increases IGF-1 levels and prevents stress susceptibility by modulating the LAP/LIP ratio.
Fig. 11 Maternal behavior increases IGF-1 levels and prevents stress susceptibility by modulating the LAP/LIP ratio.
The medial preoptic area (MPOA) is a key brain region associated with maternal behavior, and Esr1⁺ cells in the MPOA are naturally and preferentially activated during maternal behavior. To further clarify the relationship between early maternal care and adolescent stress susceptibility, the researchers used chemogenetic techniques to inhibit Esr1⁺ cell activity in the MPOA of mothers from postnatal days 0 to 8, thereby reducing early maternal behavior. They then performed 10min×2 maternal separation or control treatments, followed by the AcSD paradigm (Fig. 12k–l).
The results showed that CNO-treated mothers had fewer maternal behavior samples (Fig. 12m), offspring mPFC IGF-1 levels were reduced (Fig. 12n), LAP/LIP ratios and DRP1 mRNA and protein levels were increased (Fig. 12o), and adolescent offspring exhibited social avoidance and sucrose preference deficits, indicating that early maternal behavior loss increases adolescent stress susceptibility (Fig. 12p–q). Moreover, reduced maternal behavior did not affect offspring rearing quality. These findings underscore the critical role of early maternal behavior in regulating IGF-1 levels in the adolescent mPFC, enhancing stress recovery, and preventing depression-like behaviors (Fig. 12r).
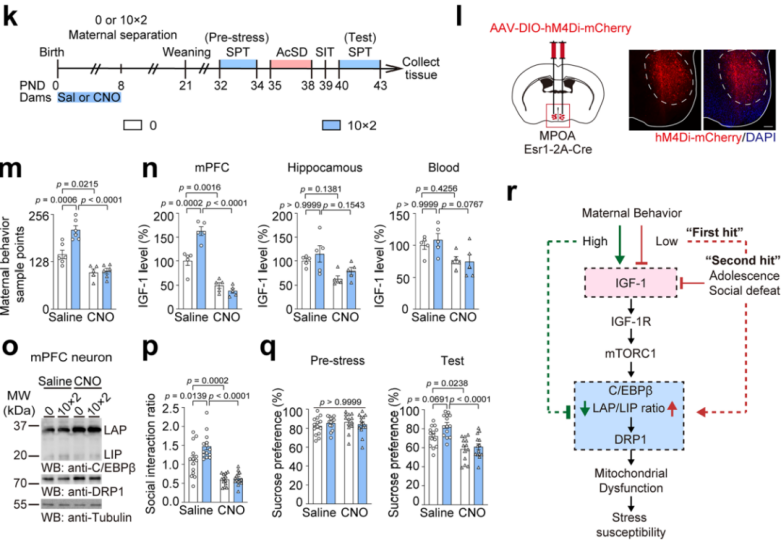
Fig. 12 Early maternal behavior loss increases adolescent stress susceptibility.
6. Microglia-Derived IGF-1 Inhibits Neuronal C/EBPβ-DRP1 Axis to Prevent Stress Susceptibility in Adolescent Mice
To identify the source of IGF-1 induced by maternal care, the researchers conducted two sets of experiments. In one set, offspring were treated with L or LnL, while in the other, mother mice were treated with saline or CNO, followed by measurements of IGF-1 levels in the offspring (Fig. 13a–c). The results showed that IGF-1 levels in the mPFC were positively correlated with changes in maternal behavior, while IGF-1 levels in the blood or hippocampus showed no such correlation (Fig. 13d–e). Immunostaining revealed high co-localization of Iba1 with Igf1 mRNA, with Igf1 mRNA levels in Iba1⁺ cells elevated in the LnL group and reduced in CNO-treated mothers, while no differences were observed in Igf1 mRNA levels in Iba1⁻ cells (Fig. 13f–g). These findings suggest that microglia are the primary source of IGF-1 in the offspring's mPFC in response to maternal behavior.
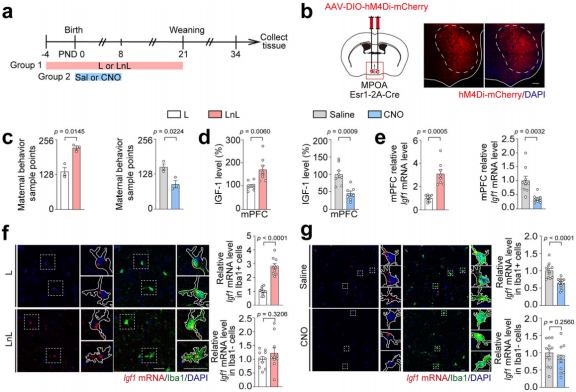
Fig. 13 Microglia are the primary source of IGF-1 in offspring responding to maternal behavior.
Microglia are the only cell type in the brain that expresses the colony-stimulating factor 1 receptor (CSF1R). Administration of the CSF1R inhibitor PLX3397 effectively depletes microglia. To investigate the role of microglia in the maternal behavior-mediated anti-depressant effects of IGF-1 in adolescent mice, PLX3397 was administered to offspring after L or LnL treatments and AcSD paradigm (Fig. 14h). The results showed that PLX3397 treatment blocked the increase in IGF-1 in the mPFC induced by LnL, as well as C/EBPβ-DRP1 activation, leading to DRP1-related mitochondrial dysfunction (Fig. 14i–n). These findings indicate that microglia depletion reduces IGF-1 levels, which in turn activates C/EBPβ's transcriptional activity on Dnm1l, causing mitochondrial dysfunction.
Behavioral tests revealed that PLX3397 treatment eliminated the protective effects of increased maternal behavior on AcSD, leading to reduced social interaction, shorter time spent in the interaction area, and impaired sucrose preference (Fig. 14o–p). Combining microglial phenotype experiments, it was demonstrated that anti-inflammatory microglia in the mPFC produce and secrete IGF-1 in response to maternal care, which helps prevent depression-like behavior by improving mitochondrial function in adolescent mice.

Fig. 14 The protective role of anti-inflammatory microglia in adolescent depression-like behavior.
To further understand the interactions between microglia and neurons, the researchers used Cx3cr1-Cre mice and AAV-MG1.2-DIO virus to regulate IGF-1 levels derived from microglia and specifically overexpressed the C/EBPβ isoform in neurons via AAV virus. Selectively inhibiting microglia-derived IGF-1 in LnL-treated offspring’s mPFC neurons and overexpressing the LIP isoform (Fig. 15a). The experimental results showed that knockdown of microglia-derived IGF-1 increased the LAP/LIP ratio and DRP1 protein expression (Fig. 15b–e), promoted mitochondrial dysfunction (Fig. 15f–g), and caused social avoidance and sucrose preference deficits. Overexpression of the LIP isoform in neurons reversed these changes and alleviated depression-like behavior (Fig. 15h–i).
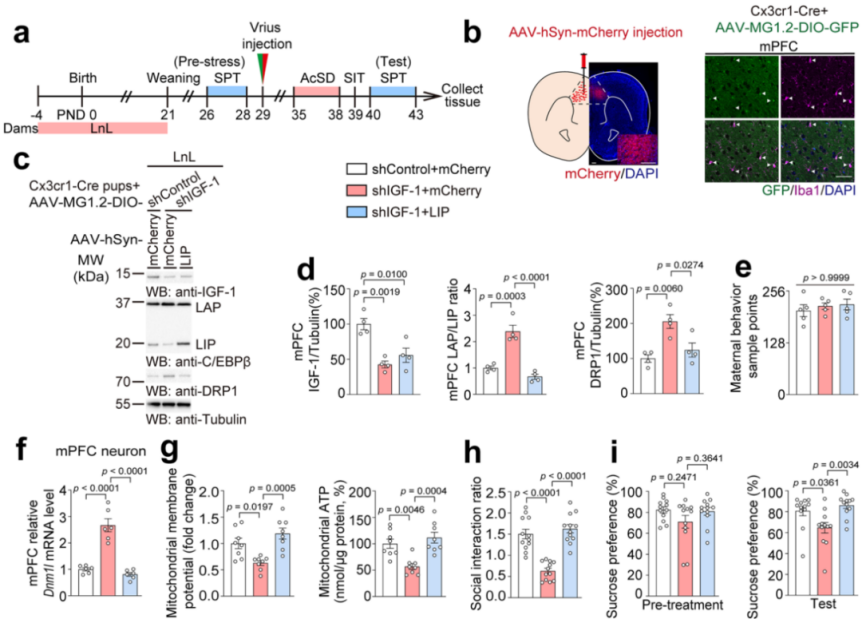 Fig. 15 Inhibition of microglia-derived IGF-1 and overexpression of the LIP isoform.
Fig. 15 Inhibition of microglia-derived IGF-1 and overexpression of the LIP isoform.
Additionally, IGF-1 was supplemented in the microglia of offspring from CNO-treated mothers, while the LAP isoform was overexpressed in mPFC neurons (Fig. 16j). The results showed that IGF-1 supplementation inhibited the LAP/LIP ratio, reduced DRP1 protein expression (Fig. 16k–n), upregulated mitochondrial MMP and ATP levels (Fig. 16o–q), and improved social avoidance and sucrose preference deficits (Fig. 16r). However, overexpression of LAP in neurons reversed these effects. These results suggest that microglial-neuronal interactions via the IGF-1-C/EBPβ-DRP1 signaling pathway play a critical role in mediating stress susceptibility.
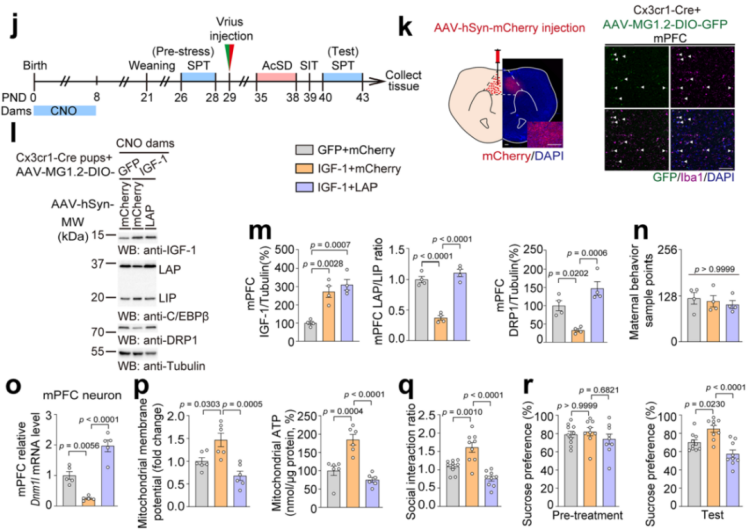
Fig. 16 Supplementing microglia-derived IGF-1 and overexpressing the LAP isoform.
7. Conclusion
This study found that maternal behavior regulates mitochondrial function and influences stress susceptibility in adolescent mice via the microglia–neuron axis, specifically through the IGF-1–C/EBPβ–DRP1 signaling pathway. It was demonstrated that social stress activates C/EBPβ in neurons of the adolescent mPFC, subsequently upregulating Dnm1l, leading to mitochondrial dysfunction and increased vulnerability to stress. In contrast, maternal behavior-induced IGF-1 inhibits this pathway and exerts a protective effect. These findings deepen our understanding of the neurobiological mechanisms underlying adolescent stress response.
Furthermore, the study confirms that early maternal care is critical in preventing depression-like behaviors during adolescence, providing a scientific foundation for early intervention in adolescent depression. The results suggest that improving early-life environments and enhancing maternal care may reduce the risk of depression in teenagers, offering important guidance for public health strategies and interventions.
All viral vectors used in this study are available from Brain Case Biotech