随着基因组测序技术的飞速发展,人们对基因组的认识不断深入,但基因组编辑能力相对滞后。CRISPR技术源于微生物免疫系统,能够实现对核酸的可编程靶向,为基因编辑带来了新契机。不过,该技术最初是为细菌的适应性免疫而进化,在应用于基础研究和临床转化时,存在脱靶效应、双链断裂(DSB)诱导的毒性以及编辑结果不可预测等问题。
2024年2月2日发表于为nature reviews molecular cell biology杂志的"CRISPR technologies for genome, epigenome and transcriptome editing"一文,详细探讨了CRISPR技术的最新进展,涵盖其在基因组编辑、表观基因组工程、转录组工程中的应用,以及这些技术在基础研究和临床治疗中的潜力与挑战。

在众多CRISPR系统中,II型CRISPR效应器Cas9凭借操作简便、可编程性强的优势,在哺乳动物基因组编辑中得到广泛应用。它借助引导RNA(gRNA)与目标DNA序列互补配对,引导Cas9蛋白结合并切割DNA,产生DSB,随后细胞通过非同源末端连接(NHEJ)、微同源介导的末端连接(MMEJ)和同源定向修复(HDR)等途径修复断裂,实现基因编辑。
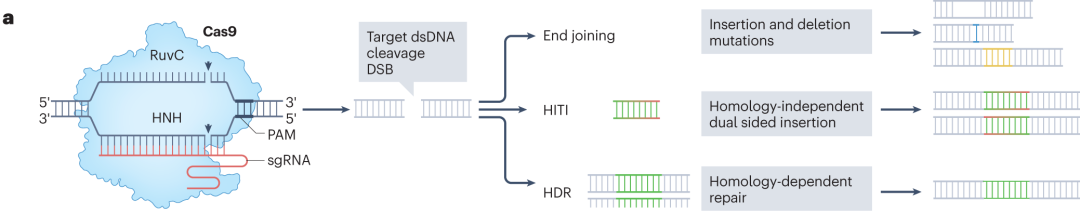
图1 DNA的损伤修复途径
1. 精确基因组编辑
1.1 碱基编辑器
碱基编辑器可在不引发DSB的情况下,对目标碱基进行精准化学修饰,主要分为胞嘧啶碱基编辑器(CBEs)和腺嘌呤碱基编辑器(ABEs)。CBEs通常由dCas9、胞嘧啶脱氨酶(如rAPOBEC1)及其他辅助蛋白构成。编辑时,dCas9在sgRNA引导下结合目标DNA序列,形成R-loop结构,暴露部分单链DNA,胞嘧啶脱氨酶特异性识别并将胞嘧啶转化为尿嘧啶,后续在DNA复制或修复过程中,尿嘧啶被细胞聚合酶识别为胸腺嘧啶,实现CG到TA的碱基转换。ABEs则是利用实验室进化的脱氧腺苷脱氨酶,将腺苷转化为肌苷,细胞聚合酶将肌苷识别为鸟苷,达成AT到GC的碱基转变。
为提升碱基编辑器性能,研究人员从多方面进行优化。调整脱氨酶与Cas9蛋白连接域的氨基酸序列,增强协同作用,减少插入缺失突变;融合UGI结构域,抑制尿嘧啶糖基化酶活性,提高编辑准确性和纯度。开发具有不同原间隔相邻基序(PAM)偏好的Cas9变体,如NG-Cas9和SpRY,拓宽编辑位点范围。通过定点突变改造脱氨酶结构域,增强对目标碱基的特异性,降低脱靶编辑频率。
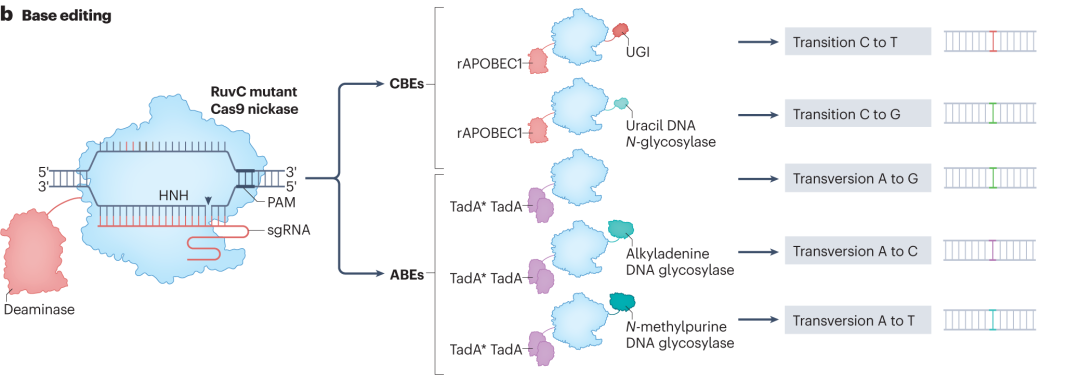
图2 碱基编辑器CBE和ABE的编辑途径
1.2 引导编辑器
引导编辑器由融合切口酶Cas9(nCas9)和逆转录酶(RT)的蛋白组件以及引导编辑向导RNA(pegRNA)组成。pegRNA不仅包含引导Cas9蛋白结合到目标位点的sgRNA序列,还携带一段编码所需DNA编辑的RNA延伸序列,作为逆转录模板。编辑过程中,nCas9在pegRNA引导下在目标DNA位点产生单链切口,pegRNA的引物结合序列与切口附近单链DNA互补配对,逆转录酶以pegRNA的延伸序列为模板进行逆转录,合成含有编辑信息的DNA单链。这条新合成的DNA单链与基因组DNA发生重组,实现精确的插入、删除或碱基替换等多种DNA修饰。
引导编辑器的优化围绕多个关键部分展开。在蛋白组件优化方面,对RT进行改造,引入特定的突变,增强其热稳定性、持续合成能力和与模板-引物复合物的结合能力,减少结合过程中的错配,提升编辑准确性和效率。在pegRNA的优化上,添加茎环等结构化RNA序列、进行化学修饰、优化pegRNA的转录启动子和终止子序列,以及调整pegRNA与Cas效应器的相互作用界面,提高pegRNA稳定性、转录效率和编辑活性。通过高通量筛选发现错配修复蛋白的关键调节作用,基于此开发出PE4和PE5等改进版本,提高编辑效率和准确性。
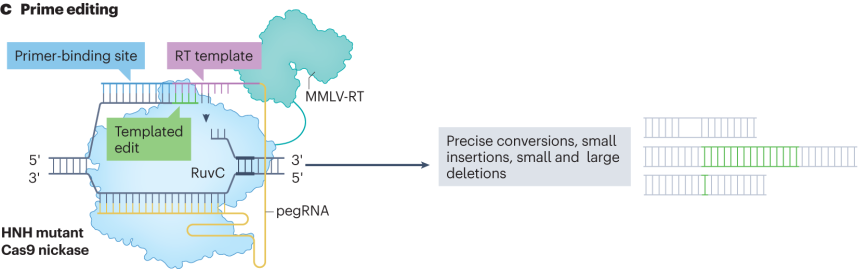
图3 引导编辑器的编辑途径
2. 大规模基因组编辑
2.1 可编程大插入
为规避DSB带来的风险,研究人员开发多种基于CRISPR技术的可编程大插入方法。dCas9与Ginβ重组酶融合理论上可实现大基因组缺失,dCas9引导Ginβ重组酶至特定基因组区域,Ginβ重组酶识别并结合特定重组序列催化DNA重组,但该方法依赖特定序列,可编程性受限。将Piggybac转座酶、Sleeping beauty转座酶与Cas9结合,是另一种大插入策略。Piggybac转座酶偏好插入TTAA位点,与Cas9结合后期望实现精准插入,但它靶向偏好宽泛,存在半随机插入问题,Sleeping beauty转座酶也有类似情况。
dCas9通过MS2发夹结构招募单链DNA退火蛋白(SSAPs),为大序列插入提供新途径。当dCas9在sgRNA引导下结合目标DNA位点形成R-loop结构时,MS2发夹结构会招募带有MS2结合结构域的SSAPs。SSAPs结合单链DNA区域,促进其与携带的双链DNA模板进行退火和重组,实现大序列的插入。这种方法能够有效减少非预期的末端连接编辑结果,提高插入的准确性和稳定性。
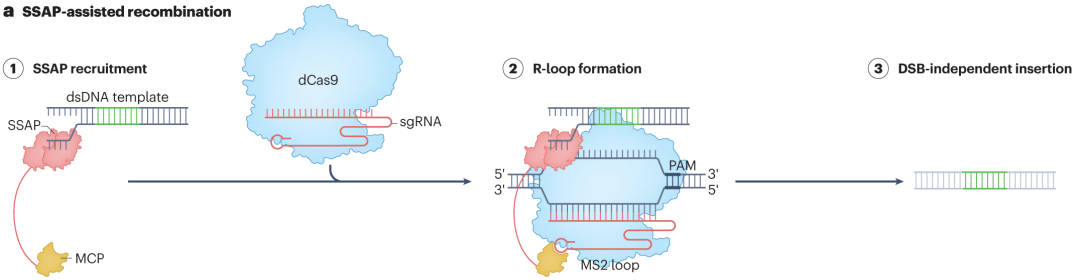
图4 SSAP关联的可编程大插入
CRISPR相关转座酶(CASTs)系统是备受关注的大插入技术。在细菌中,CASTs系统利用RNA引导的转座作用,可高效将Tn7样转座子插入目标位点。但在哺乳动物细胞中,其整合效率较低。目前,研究人员正通过结构引导诱变和定向进化等技术对其优化,期望提高在哺乳动物细胞中的整合效率,以更好地应用于基因治疗和细胞工程等领域。
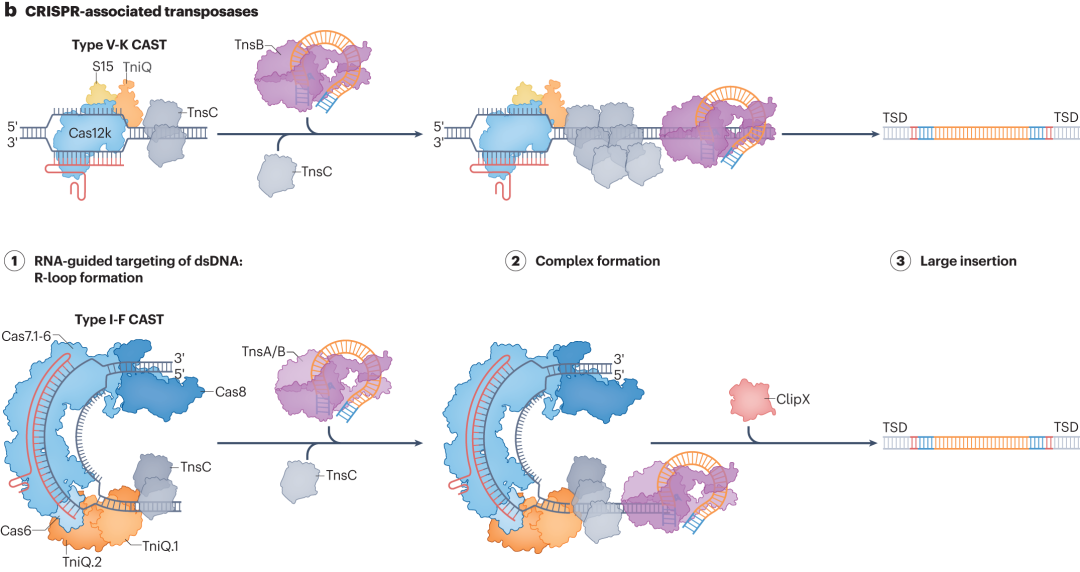
图5 CASTs关联的可编程大插入
基于引导编辑的PASTE和TwinPE knock-in技术为大序列插入带来突破。PASTE技术利用pegRNA引导编辑器在目标位点插入一段特定的DNA序列,作为后续整合的着陆点。然后,丝氨酸整合酶识别并结合该着陆位点和携带目标基因的双链DNA供体上的相应位点,催化两者之间的重组反应,将供体DNA整合到目标位置,可实现高达36kb的cargo插入。TwinPE knock-in则依赖两个pegRNAs和整合酶,两个pegRNAs分别引导编辑器在目标位点的两侧产生单链切口,形成可供整合酶识别的位点。整合酶将携带目标基因的供体DNA准确地整合到两个切口之间,实现大序列的精确插入。
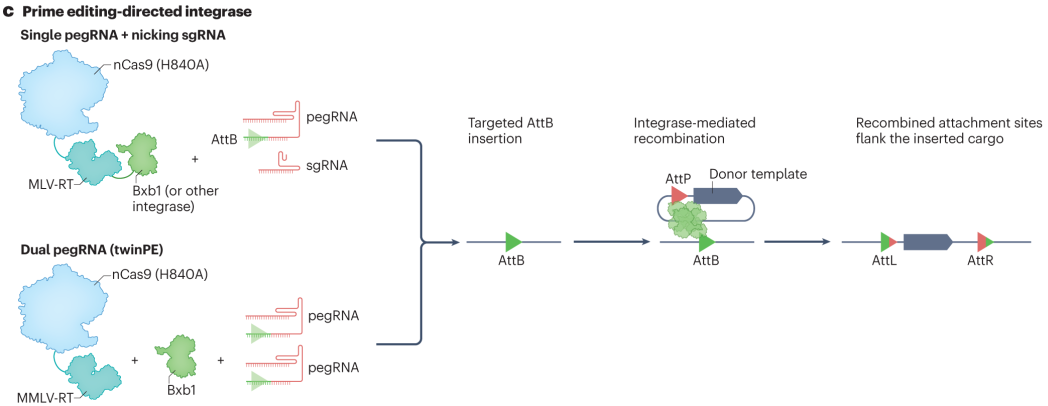
图6 PASTE和TwinPE关联的可编程大插入
2.2 大基因组缺失
传统利用双sgRNA诱导Cas9介导的DSB实现大基因组缺失的方法存在诸多问题,修复过程随机,可能导致删除大小不可预测,还可能引发染色体重组等异常事件。HOPE、PRIME-Del、双向引导编辑(Bi-PE)等方法是引导编辑技术在大基因组缺失应用中的具体体现。HOPE方法通过巧妙设计pegRNAs,使合成的flap能够精确互补,提高了编辑的准确性,可实现小的插入、删除以及精确的大基因组缺失。PRIME-Del和Bi-PE则进一步拓展了引导编辑的应用范围,实现更大范围的插入(可达30bp)和删除(可达10kb),在处理复杂的DNA重排,如倒位等方面也表现出色。
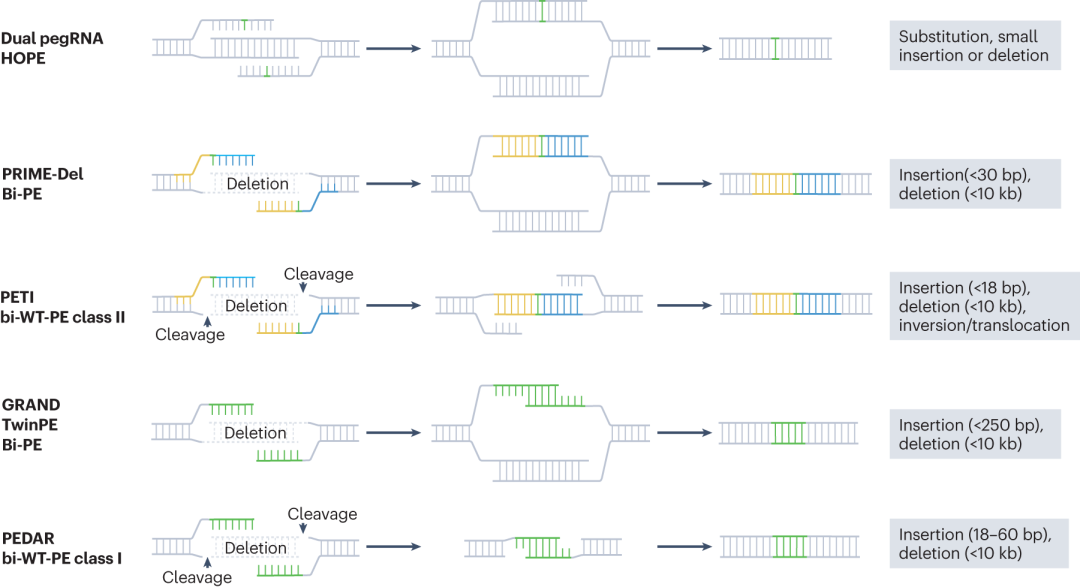
图7 可编程的大基因组缺失
2.3 可编程逆转录转座子
目前多数大基因组序列插入或删除技术依赖DNA插入模板,限制了RNA递送的应用。基于逆转录转座子的方法为此提供了新思路。R2非长末端重复逆转录转座子是其中的典型代表,它具有独特的生物学特性,能够结合目标DNA、具备核酸内切酶活性并进行逆转录,最终将自身插入到28S核糖体DNA位点。
在其作用过程中,R2非长末端重复逆转录转座子首先识别并结合到目标DNA序列上,然后利用自身的核酸内切酶活性在目标位点产生一个切口,以自身携带的RNA为模板进行逆转录。最后,将合成的DNA整合到目标位点,完成转座过程。然而,其RNA模板插入活性较低。研究人员正在积极探索新的R2同源物,并通过基因工程手段改造现有R2转座子,期望提升其活性,使其在临床应用中发挥更大作用。
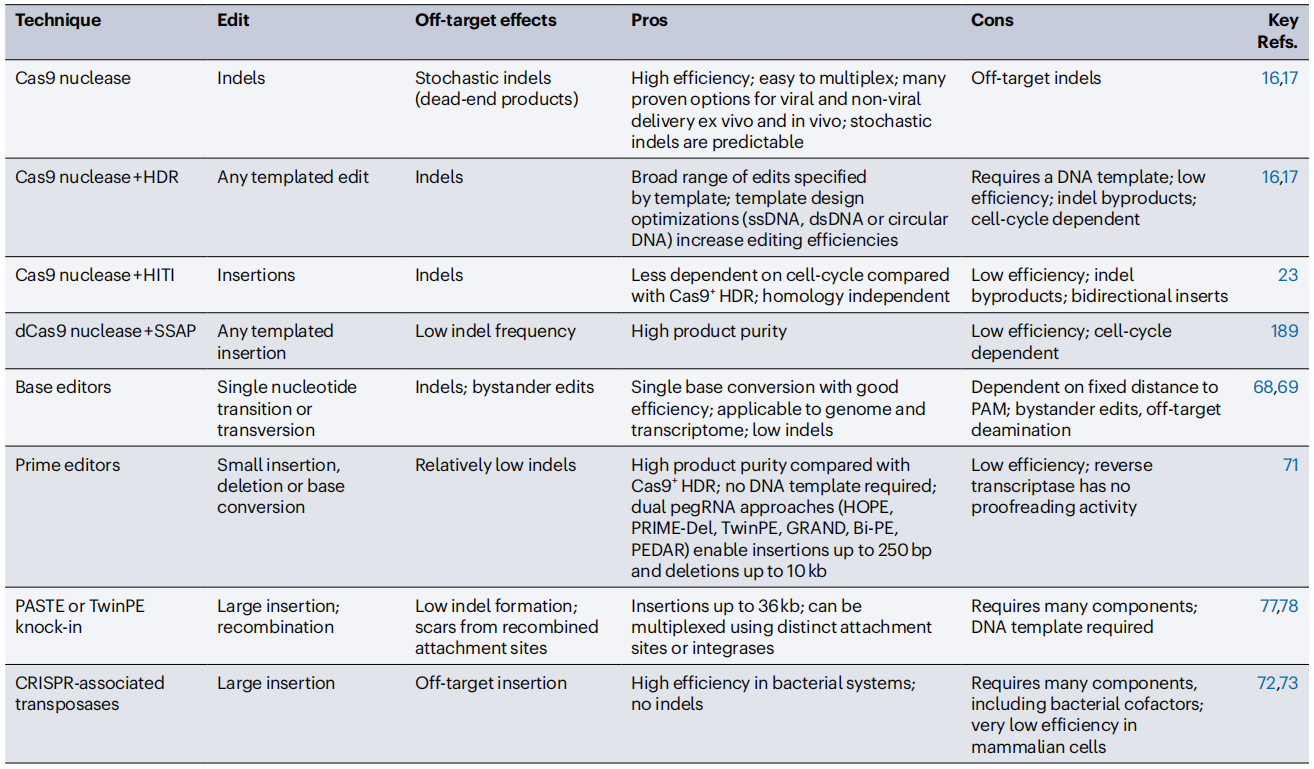
表1 可编程编辑技术与基因插入技术的比较
1. 转录的瞬时调控
CRISPR-Cas系统可在不改变基因组序列的情况提下实现转录的瞬时调控,主要通过将转录激活或抑制结构域与催化失活的Cas9(dCas9)融合来实现。在CRISPR激活(CRISPRa)技术中,第一代CRISPRa将dCas9与单个转录激活结构域(如VP64)融合,dCas9-VP64融合蛋白在sgRNA引导下结合到目标基因的启动子或增强子区域时,VP64结构域能够招募细胞内的转录相关因子,如RNA聚合酶II等,促进转录起始复合物的形成,实现转录上调。
随着研究深入,第二代CRISPRa技术不断涌现。CRISPRa v2 SunTag利用重复肽阵列SunTag招募多个VP64激活结构域,显著增强转录激活效果。协同激活介质(SAM)CRISPRa在sgRNA支架中引入两个MS2适体,招募p65和HSF1激活结构域,进一步提高转录激活效率。将VP64-p65-Rta(VPR)串联融合到dCas9,简化系统组成的同时提高转录激活效率。
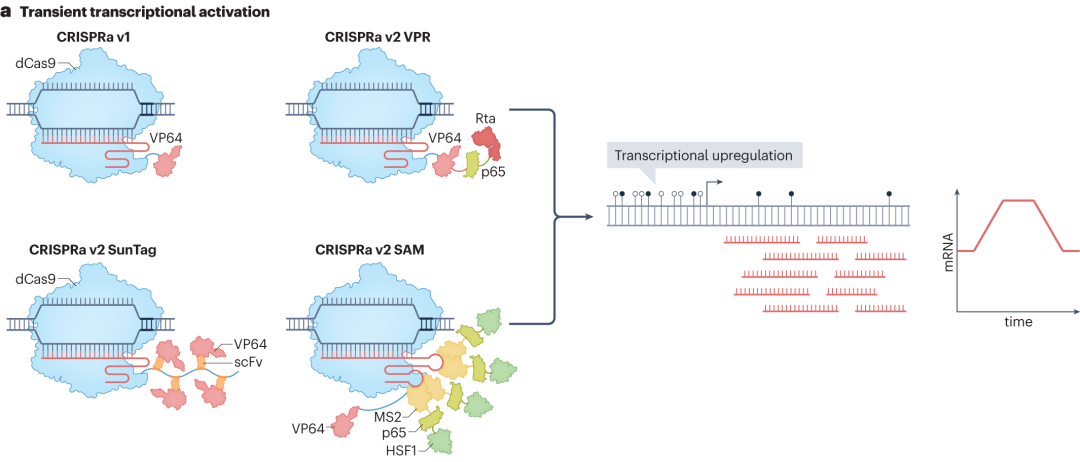
图8 CRISPRa的瞬时转录激活
在CRISPR抑制(CRISPRi)技术中,第一代CRISPRi将dCas9与锌指蛋白10(ZNF10)的KRAB抑制结构域融合,KRAB结构域招募转录抑制相关蛋白,改变染色质结构,阻碍RNA聚合酶与启动子结合,实现转录下调。后续的CRISPRi系统通过替换更有效的抑制结构域,如用锌指印记3(ZIM3)的KRAB结构域替换ZNF10的KRAB结构域,以及添加MeCP2和SID4x等抑制结构域,增强转录抑制效果。
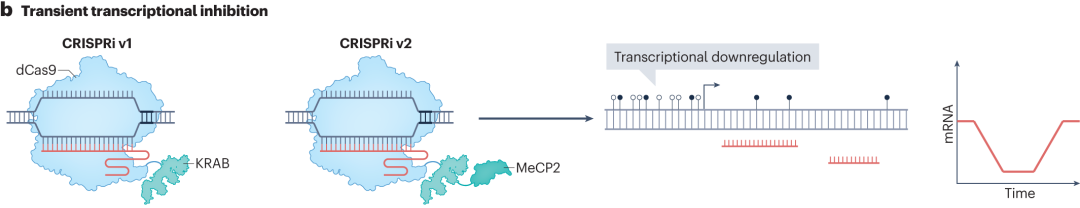
图9 CRISPRi的瞬时转录抑制
2. 转录的稳定调控
CRISPRa和CRISPRi虽然实现可编程的转录组工程,但需要持续表达dCas9融合蛋白来维持转录控制,在临床治疗等实际应用中存在一定局限性。为此,研究人员开发了CRISPRoff和CRISPRon技术。
CRISPRon是将优化的dCas9与TET1 DNA去甲基酶的催化结构域和MS2 sgRNA支架融合。催化DNA去甲基化反应,促进转录相关因子的结合,从而激活转录。此外,将dCas9与超活性组蛋白激酶(MSK1)或组蛋白乙酰化酶(p300/CBP)融合,可分别实现靶组蛋白的磷酸化或乙酰化。组蛋白的磷酸化和乙酰化修饰能够改变染色质的结构和功能,使基因更容易被转录相关因子识别和结合,进而激活基因表达。
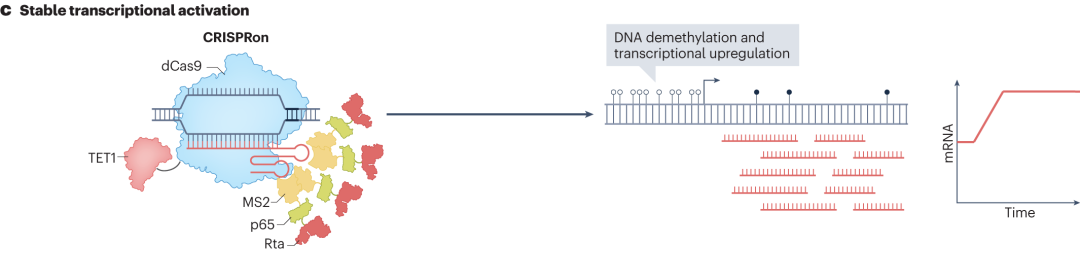
图10 CRISPRon的稳定转录激活
CRISPRoff则是由KRAB、Dnmt3A和Dnmt3L蛋白结构域与dCas9融合而成。在sgRNA的引导下结合到目标基因组区域时,Dnmt3A和Dnmt3L蛋白结构域能够对该区域的DNA进行甲基化修饰。通过瞬时表达CRISPRoff,可在基因组特定区域建立稳定的DNA甲基化,实现转录抑制,这种抑制在细胞分裂和干细胞分化过程中稳定维持。
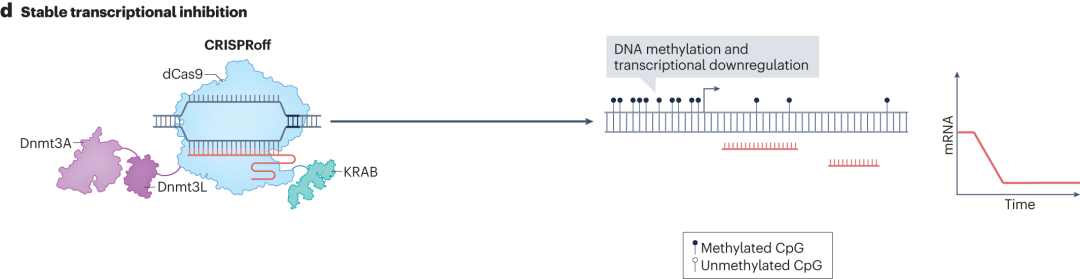
图11 CRISPRoff的稳定转录抑制
1. RNA敲低
RNA靶向的CRISPR核酸酶的发现为转录组工程开辟了全新的领域。Cas13a是一种来自II类VI型CRISPR系统的单效应核酸酶,通过可编程的gRNA特异性识别并结合靶RNA序列,然后Cas13a的核酸酶结构域对靶RNA进行切割,实现对靶基因表达的下调。但Cas13a切割靶RNA后,会产生非特异性切割附近RNA的旁切活性。这种旁切活性可能导致细胞内大量非靶RNA被降解,影响细胞的正常生理功能。后续研究发现的Cas13b、Cas13d等同源物虽敲低效率更高,但同样存在旁切活性问题。
为解决这一问题,研究人员开发具有工程化核酸酶突变的RfxCas13d变体,通过对RfxCas13d的核酸酶结构域进行定点突变,改变其空间构象和底物结合特异性,有效减少非特异性RNA结合和旁切活性。研究人员还发现了小型Cas13核酸酶Cas13X,优化其结构和功能后,在保持高效RNA敲低能力的同时,极大降低旁切活性。
此外,Cas7-11是一种新型的RNA靶向CRISPR核酸酶,它属于I类III-E型CRISPR系统。它能够切割靶RNA且无旁切活性,降低潜在的细胞毒性。此外,III型效应复合物如CRISPR-Csm也可用于RNA敲低。在斑马鱼细胞、大肠杆菌和哺乳动物细胞中均表现良好,为RNA敲低技术在不同生物模型中的应用提供更多选择。
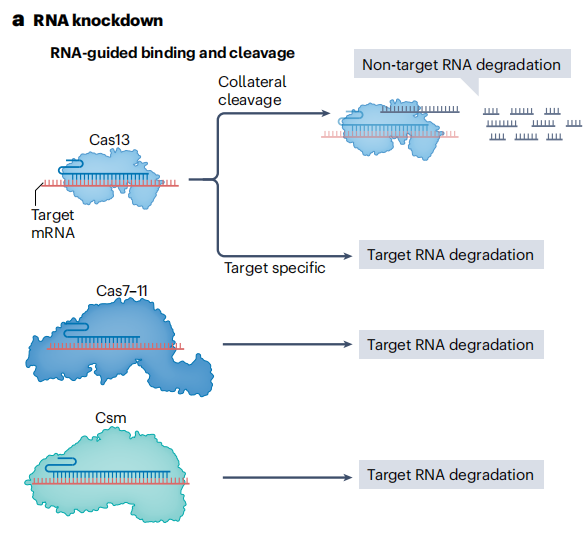
图12 RNA敲低
2. RNA碱基编辑
基于ADAR家族脱氨酶的RNA编辑工具,能够在双链RNA(dsRNA)底物上催A-to-I编辑。ADAR(Adenosine Deaminase Acting on RNA)家族脱氨酶可以识别并作用于dsRNA中的腺苷,将其转化为肌苷,而肌苷在翻译过程中会被识别为鸟苷,实现RNA水平的碱基转换。
实现ADAR-based编辑的方法多种多样。将ADAR与gRNA共价连接,通过MS2或BoxB结构域将工程化的ADAR与gRNA高亲和力结合,招募内源性ADAR酶来实现RNA编辑,这种方法利用细胞自身的ADAR酶资源,减少外源性蛋白的引入,降低潜在的免疫原性和细胞毒性。REPAIR技术将dCas13b与超活性ADAR2变体融合,能够在特定RNA碱基上引入编辑,校正疾病相关突变。但初代版本的REPAIR存在脱靶编辑问题,超活性ADAR2在非靶位点也可能发生编辑作用,影响正常基因的表达。通过对ADAR2进行理性蛋白质工程改造,开发出REPAIR v2,显著减少了脱靶编辑。
为实现C-to-U编辑,研究人员通过进化ADAR2使其具有胞嘧啶脱氨酶活性,并与dCas13融合,开发出了RESCUE工具。RESCUE通过在ADAR2中引入特定的突变,使其能够识别并作用于胞嘧啶,将其转化为尿嘧啶。为了减少脱靶编辑,RESCUE还引入了额外的突变,降低了ADAR2对非靶位点的活性。此外,利用更紧凑的EcCas6e蛋白替代dCas13,开发出了高效的RNA碱基编辑器ceRBE,在体内编辑效率和减少转录组脱靶效应方面表现优异。

图13 RNA碱基编辑
3. RNA修饰
将dCas13与截短的METTL3甲基转移酶结构域融合,能够在特定RNA转录本上安装N⁶-甲基腺苷(m⁶A)修饰。m⁶A修饰在调节转录本丰度和可变剪接中发挥重要作用,它可以影响RNA的稳定性、翻译效率以及细胞内定位等过程。通过这种融合蛋白,研究人员可以在特定的RNA分子上引入m⁶A修饰,进而研究其对基因表达调控的影响。
REMOVER工具利用CRISPR-Cas13d实现了对N¹-甲基腺苷(m¹A)位点的靶向去甲基化。有助于了解m¹A在调节RNA命运和基因表达中的作用机制。通过将脂肪量和肥胖相关蛋白(FTO)或AlkB5同源物5(ALKBH5)等RNA去甲基酶与Cas13融合,实现了对m⁶A的可编程位点特异性去除,为研究m⁶A修饰的动态变化和功能提供了有力的工具。随着RNA修饰编辑器的不断优化,其在转录组表型筛选和解析特定修饰在不同细胞通路中的作用方面将发挥更大的潜力。
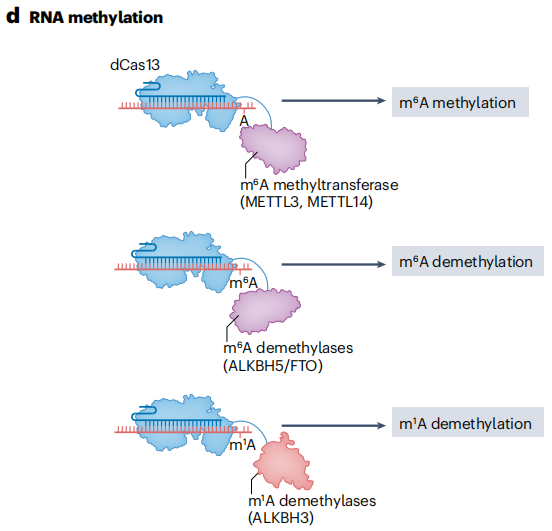
图14 RNA甲基化修饰
CRISPR技术凭借其可编程性和强大活性,在临床治疗领域展现出巨大的应用潜力,广泛涵盖了体外细胞工程和体内基因校正等多个重要方面。
在癌症免疫治疗方面,多项临床试验正在开展。通过体外使用Cas9核酸酶敲除T细胞中的免疫抑制基因,如程序性死亡受体1(PD-1),可增强T细胞的活性,使其能够更有效地识别和杀伤肿瘤细胞。敲除天然T细胞受体(TCR),并引入工程化的TCR或嵌合抗原受体(CAR),能够使T细胞特异性地识别肿瘤细胞表面的抗原,大大提高T细胞对肿瘤细胞的靶向性和杀伤能力,部分接受治疗后病情得到了缓解。
在治疗单基因血液疾病方面,CRISP技术也展现出了巨大的潜力。镰状细胞病和输血依赖性β-地中海贫血与血红蛋白基因的突变有关。通CRISPR-Cas9编辑造血干细胞(HSCs),可以破坏相关基因的调控元件,如BCL11A,可促进胎儿血红蛋白的代偿性表达,改善患者的贫血症状。
在体内基因校正方面,CRISPR技术主要应用于治疗一些组织相对易于递送CRISPR核酸酶的遗传疾病。如通过脂质纳米颗粒(LNPs)递送Cas9 mRNA和sgRNA治疗转甲状腺素蛋白淀粉样变性,可敲除肝脏中的转甲状腺素蛋白基因,降低有毒蛋白浓度,减轻器官损害。利用LNPs递送Cas9敲除激肽释放酶基因,可缓解遗传性血管性水肿的症状。
CRISPR技术在基因组、表观基因组和转录组编辑领域取得显著进展,开发出多种编辑工具,推动了基础研究和临床治疗的发展,在癌症免疫治疗、单基因血液疾病治疗等方面潜力巨大。然而,该技术在临床应用中面临转基因递送、免疫原性、编辑准确性和安全性等挑战。未来需结合机器学习和测序技术优化编辑工具,开发新型递送载体,深入探索新基因靶点,并将其与细胞状态响应工具结合,同时重视伦理监管,以推动CRISPR技术在医学和生物技术领域发挥更大作用。
参考文献
Villiger L, Joung J, Koblan L, Weissman J, Abudayyeh OO, Gootenberg JS. CRISPR technologies for genome, epigenome and transcriptome editing. Nat Rev Mol Cell Biol. 2024 Jun;25(6):464-487. doi: 10.1038/s41580-023-00697-6. Epub 2024 Feb 2. Erratum in: Nat Rev Mol Cell Biol. 2024 Jun;25(6):510. doi: 10.1038/s41580-024-00745-9. PMID: 38308006.



地址:-
地址:-
