IF=85 Cutting-edge Review | CRISPR’s Three-Dimensional Breakthrough: A Comprehensive Overview of Genome, Epigenome, and Transcriptome Editing
Release time:2025-06-05 11:15:45
With the rapid advancement of genome sequencing technologies, our understanding of the genome has deepened significantly. However, genome editing capabilities have lagged behind. Originating from microbial immune systems, CRISPR technology enables programmable targeting of nucleic acids, opening new avenues for gene editing. Nevertheless, as CRISPR evolved primarily for adaptive immunity in bacteria, its application in basic research and clinical translation faces several challenges, including off-target effects, toxicity induced by double-strand breaks (DSBs), and unpredictable editing outcomes. On February 2, 2024, Nature Reviews Molecular Cell Biology published the article "CRISPR technologies for genome, epigenome and transcriptome editing", which provides an in-depth discussion of the latest developments in CRISPR technologies. The review covers applications in genome editing, epigenome engineering, and transcriptome modulation, while also examining the potential and challenges of these approaches in both fundamental research and clinical therapies.
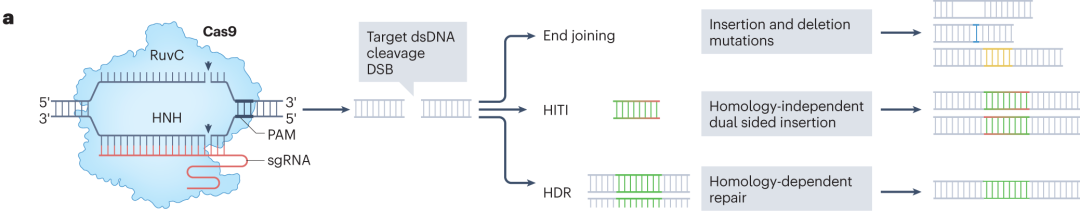
Among the various CRISPR systems, the type II CRISPR effector Cas9 has become widely used in mammalian genome editing due to its simplicity and high programmability. Cas9 is guided by a guide RNA (gRNA) that pairs with a complementary DNA sequence, directing the Cas9 protein to bind and cleave the DNA. This cleavage induces a DSB, which is subsequently repaired by the cell through pathways such as non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), or homology-directed repair (HDR), thereby enabling genome editing.
Figure 1. DNA Damage Repair Pathways
1. Precise Genome Editing
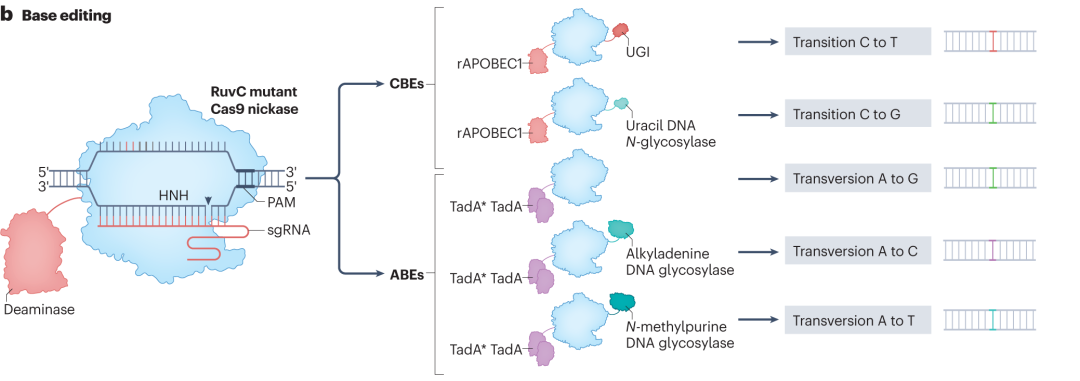
1.1 Base Editors
Base editors enable precise chemical modification of target nucleotides without inducing DSBs, and are mainly categorized into cytosine base editors (CBEs) and adenine base editors (ABEs).CBEs typically consist of a catalytically dead Cas9 (dCas9), a cytidine deaminase (e.g., rAPOBEC1), and additional auxiliary proteins. Guided by sgRNA, dCas9 binds the target DNA sequence and forms an R-loop, exposing a segment of single-stranded DNA. The cytidine deaminase specifically recognizes and converts cytosine (C) to uracil (U). During DNA replication or repair, uracil is recognized by cellular polymerases as thymine (T), thereby achieving a C•G to T•A base conversion.ABEs, on the other hand, use laboratory-evolved deoxyadenosine deaminases to convert adenine (A) to inosine (I), which is interpreted by the DNA polymerase as guanine (G), resulting in an A•T to G•C substitution.
To enhance the performance of base editors, researchers have made several optimizations: Modifying the amino acid linkers between deaminases and Cas9 to improve synergy and reduce insertion–deletion mutations (indels);Fusing uracil glycosylase inhibitor (UGI) domains to suppress the activity of uracil-DNA glycosylase, thereby improving editing precision and purity;Engineering Cas9 variants such as NG-Cas9 and SpRY with altered PAM recognition preferences, thereby expanding the editable target range;Performing site-directed mutagenesis on deaminase domains to enhance substrate specificity and minimize off-target effects.
Figure 2. Editing Mechanisms of CBE and ABE
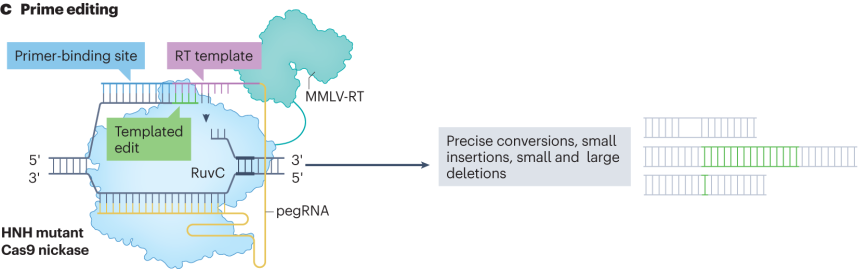
1.2 Prime Editors
Prime editors are composed of a protein component formed by the fusion of nickase Cas9 (nCas9) and reverse transcriptase (RT), along with a prime editing guide RNA (pegRNA).The pegRNA not only contains the sgRNA sequence that guides the Cas9 protein to the target site, but also carries an RNA extension sequence encoding the desired DNA edit, which serves as a reverse transcription template.
During the editing process, nCas9, guided by the pegRNA, generates a single-strand nick at the target DNA site. The primer binding site (PBS) of the pegRNA pairs with the nearby single-stranded DNA, and the reverse transcriptase uses the extension sequence of the pegRNA as a template to synthesize a new DNA strand containing the desired edit. This newly synthesized DNA strand undergoes recombination with the genomic DNA, enabling precise DNA modifications such as insertions, deletions, or base substitutions.
The optimization of prime editors focuses on several key components. In terms of protein optimization, mutations are introduced into the reverse transcriptase to enhance its thermal stability, processivity, and binding ability to the template-primer complex, thereby reducing mismatches during binding and improving editing accuracy and efficiency.
Regarding pegRNA optimization, structured RNA elements such as stem-loops are added, chemical modifications are applied, transcription promoters and terminators of the pegRNA are optimized, and the interaction interface between pegRNA and the Cas effector is adjusted, all aiming to improve pegRNA stability, transcription efficiency, and editing activity.
High-throughput screening has revealed the key regulatory roles of mismatch repair proteins, based on which improved versions such as PE4 and PE5 have been developed to enhance editing efficiency and accuracy.
Figure 3. Editing Mechanism of Prime Editors
2. Large-Scale Genome Editing
2.1 Programmable Large Insertions
To avoid the risks associated with DSBs, researchers have developed various CRISPR-based strategies for programmable large insertions. A fusion of dCas9 with Ginβ recombinase can theoretically enable large genomic deletions. dCas9 directs Ginβ recombinase to specific genomic regions, where Ginβ recognizes and binds specific recombination sequences to catalyze DNA recombination. However, this method relies on the presence of defined sequences, limiting its programmability.
Another strategy involves combining PiggyBac transposase or Sleeping Beauty transposase with Cas9. PiggyBac transposase preferentially inserts at TTAA sites. When combined with Cas9, it is expected to achieve targeted insertions. However, due to its broad target preference, semi-random insertion events may still occur—a similar issue is observed with Sleeping Beauty transposase.
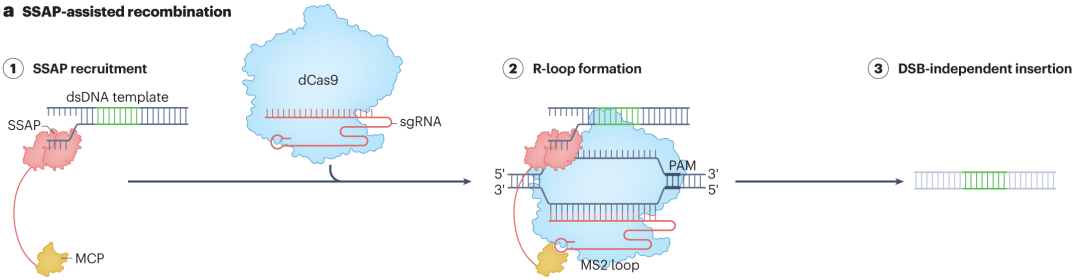
A novel approach uses dCas9 to recruit single-stranded DNA annealing proteins (SSAPs) via MS2 hairpin structures, offering a new method for large-sequence insertions. When dCas9 binds to the target DNA site under sgRNA guidance and forms an R-loop, the MS2 hairpin structure recruits SSAPs containing MS2-binding domains. SSAPs bind to single-stranded DNA regions and promote annealing and recombination with double-stranded DNA templates carrying the desired insert.This strategy effectively reduces unintended end-joining outcomes and improves the precision and stability of large-sequence insertions.
Figure 4. SSAP-Associated Programmable Large Insertions
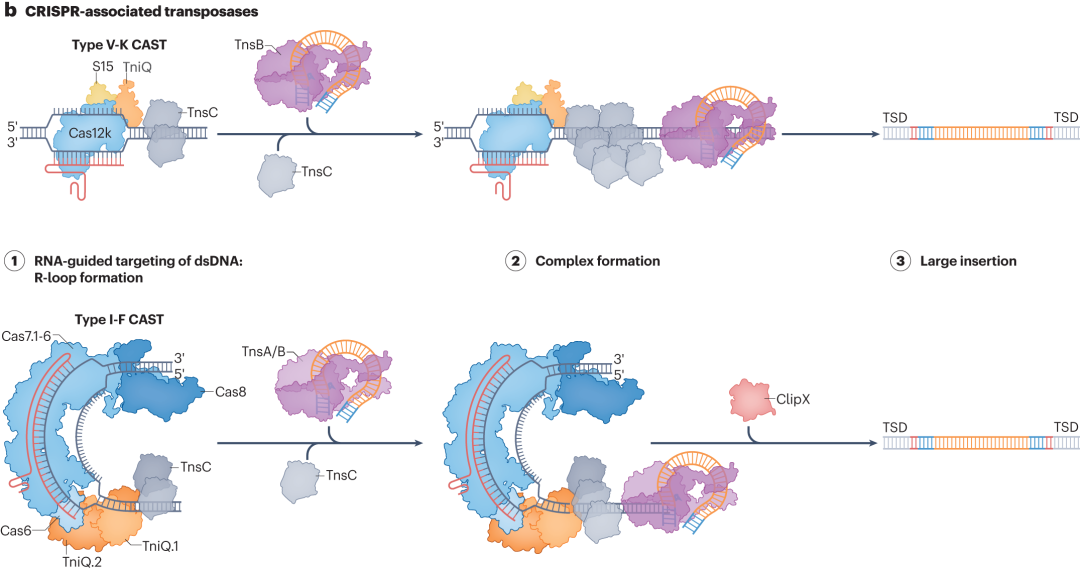
CRISPR-associated transposase (CASTs) systems are an emerging and highly anticipated technology for large DNA insertions.In bacteria, CASTs utilize RNA-guided transposition to efficiently insert Tn7-like transposons into target sites. However, in mammalian cells, their integration efficiency remains relatively low.Currently, researchers are optimizing these systems using structure-guided mutagenesis and directed evolution, aiming to improve their integration efficiency in mammalian systems for broader applications in gene therapy and cell engineering.
Figure 5. CASTs-Associated Programmable Large Insertions
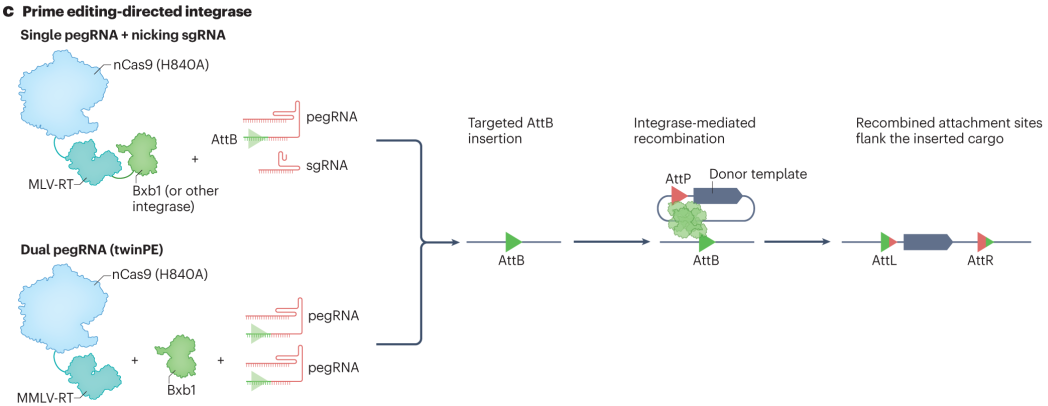
Prime editor–based technologies such as PASTE and TwinPE knock-in represent breakthroughs in large-sequence insertion.The PASTE system uses a pegRNA to guide the prime editor to insert a specific DNA landing pad at the target site. A serine integrase then recognizes and binds to the landing site and a corresponding site on a double-stranded donor DNA carrying the gene of interest, catalyzing a recombination event that integrates the donor DNA into the target locus—allowing insertions of cargo sequences up to 36 kb.
TwinPE knock-in, on the other hand, utilizes two pegRNAs and an integrase. Each pegRNA guides the editor to generate a single-strand nick on either side of the target site, creating two sites recognizable by the integrase. The integrase then precisely integrates the donor DNA between the two nicks, enabling accurate insertion of large DNA sequences.
Figure 6. PASTE and TwinPE-Associated Programmable Large Insertions
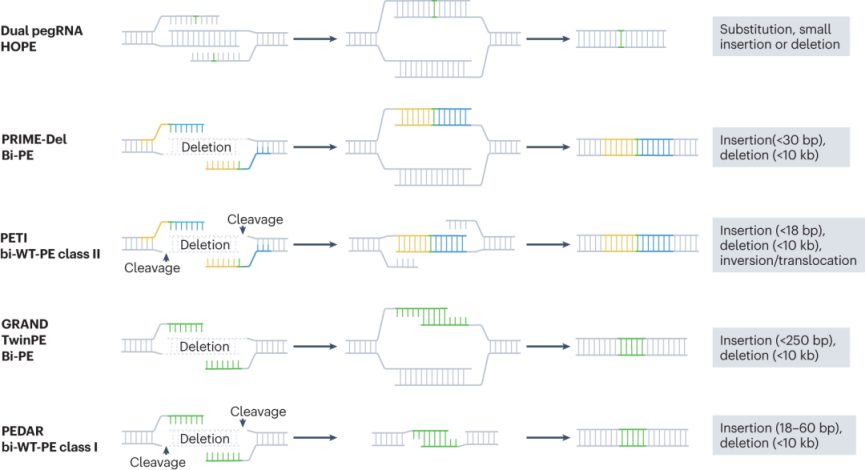
2.2 Large Genomic Deletions
Traditional methods for inducing large genomic deletions using Cas9-mediated DSBs guided by dual sgRNAs face numerous challenges. The repair process is random, which can lead to unpredictable deletion sizes and may also cause abnormal events such as chromosomal rearrangements. Technologies like HOPE, PRIME-Del, and bidirectional prime editing (Bi-PE) represent the application of prime editing in large genomic deletions. The HOPE method improves editing precision by designing pegRNAs in such a way that the synthesized DNA flaps can accurately anneal, enabling precise small insertions, deletions, and large genomic deletions. PRIME-Del and Bi-PE further expand the application scope of prime editing, enabling broader-range insertions (up to 30 bp) and deletions (up to 10 kb), and showing promising performance in addressing complex DNA rearrangements, such as inversions.
Figure 7. Programmable Large Genomic Deletions
2.3 Programmable Retrotransposons
Currently, most techniques for inserting or deleting large genomic sequences rely on DNA donor templates, which limits the applicability of RNA-based delivery. Retrotransposon-based methods offer new strategies to address this limitation.A typical example is the R2 non-long terminal repeat (non-LTR) retrotransposon, which exhibits unique biological properties—it can bind to target DNA, possesses endonuclease activity, and is capable of reverse transcription, ultimately integrating itself into the 28S ribosomal DNA locus. During this process, the R2 retrotransposon first recognizes and binds to the target DNA sequence. It then uses its endonuclease activity to generate a nick at the target site and performs reverse transcription using its own RNA as a template. Finally, the synthesized DNA is integrated into the target site, completing the transposition process. However, the RNA template–based insertion efficiency of R2 retrotransposons remains low. Researchers are actively exploring new R2 homologs and applying genetic engineering strategies to modify existing R2 retrotransposons, with the aim of enhancing their activity and realizing greater potential for clinical applications.
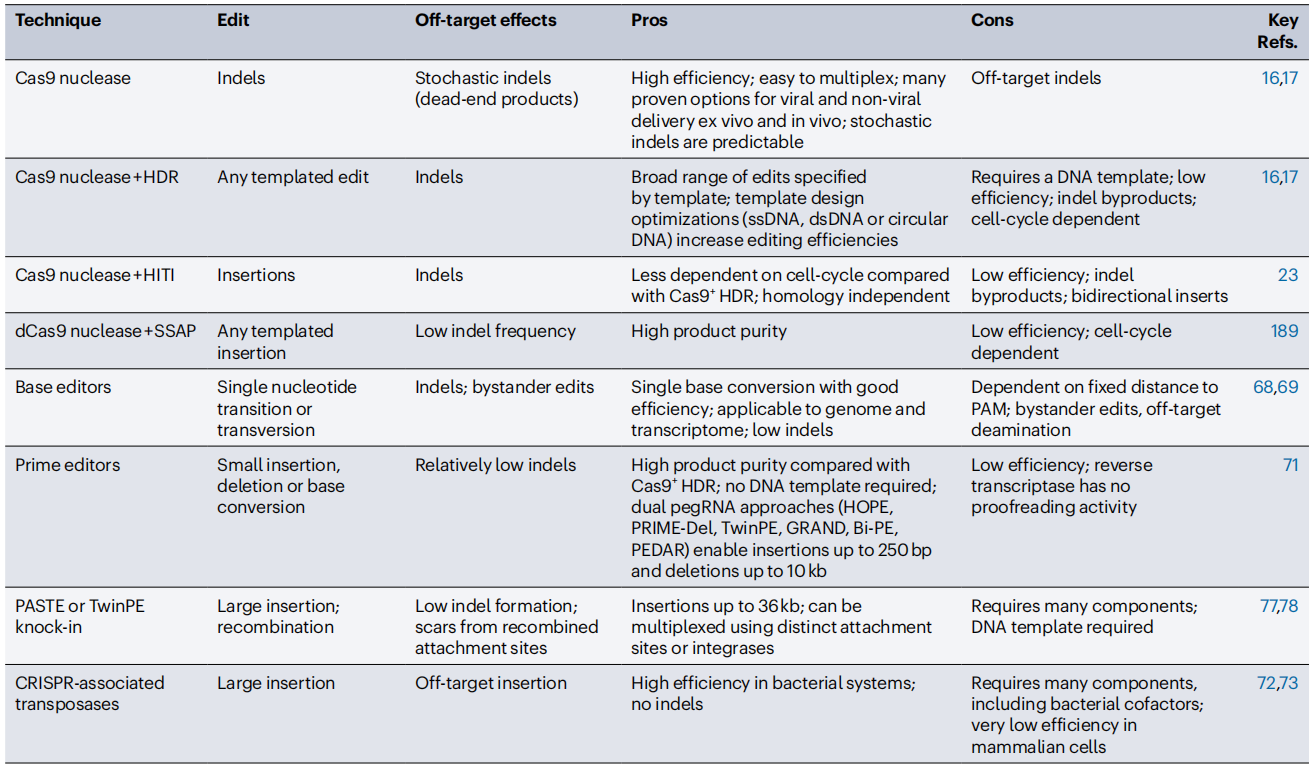
Table 1. Comparison of Programmable Editing and Gene Insertion Technologies
II. Epigenome Engineering
1. Transient regulation of transcription
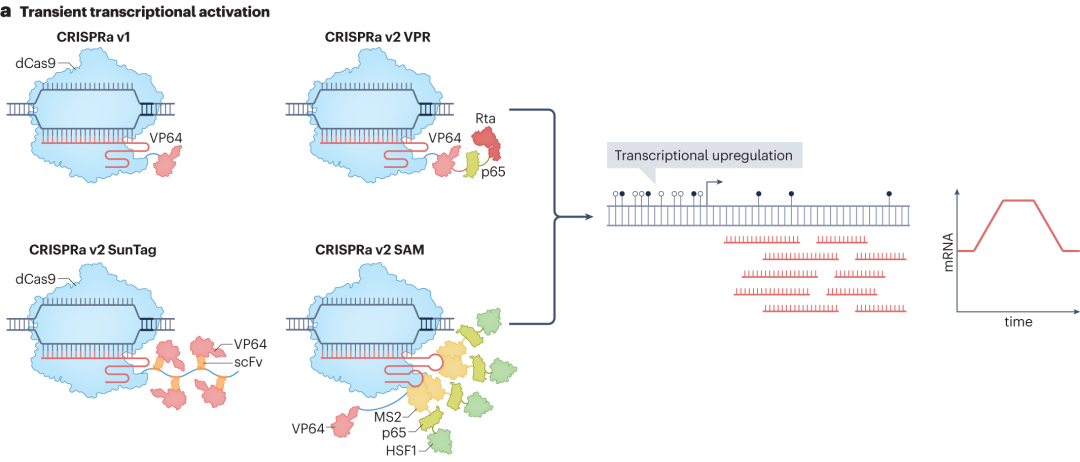
The CRISPR-Cas system can achieve transient regulation of transcription without altering the genomic sequence, primarily by fusing transcriptional activation or repression domains with catalytically inactive Cas9 (dCas9). In CRISPR activation (CRISPRa) technology, the first-generation CRISPRa fuses dCas9 with a single transcriptional activation domain (such as VP64). When the dCas9-VP64 fusion protein is guided by sgRNA to bind to the promoter or enhancer region of a target gene, the VP64 domain can recruit intracellular transcription-related factors, such as RNA polymerase II, to promote the formation of the transcription initiation complex and achieve transcriptional upregulation.
As research progresses, second-generation CRISPRa technologies continue to emerge. CRISPRa v2 SunTag utilizes a repeated peptide array, SunTag, to recruit multiple VP64 activation domains, significantly enhancing the transcriptional activation effect. The synergistic activation mediator (SAM) CRISPRa introduces two MS2 aptamers into the sgRNA scaffold to recruit activation domains such as p65 and HSF1, further improving transcriptional activation efficiency. The system that fuses VP64-p65-Rta (VPR) in tandem to dCas9 simplifies the system composition while enhancing transcriptional activation efficiency.
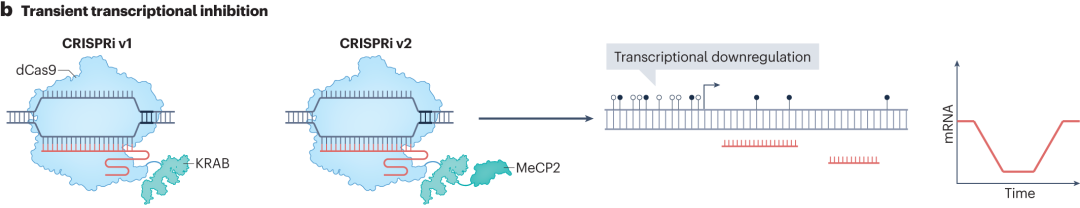
In CRISPR interference (CRISPRi) technology, the first-generation CRISPRi fuses dCas9 with the KRAB repression domain from zinc finger protein 10 (ZNF10). The KRAB domain recruits transcriptional repression-related proteins, alters chromatin structure, and blocks RNA polymerase from binding to the promoter, thereby achieving transcriptional downregulation. Subsequent CRISPRi systems have enhanced transcriptional repression by replacing the KRAB domain from ZNF10 with more effective ones, such as the KRAB domain from zinc finger imprinted 3 (ZIM3), and by adding other repressive domains such as MeCP2 and SID4x.
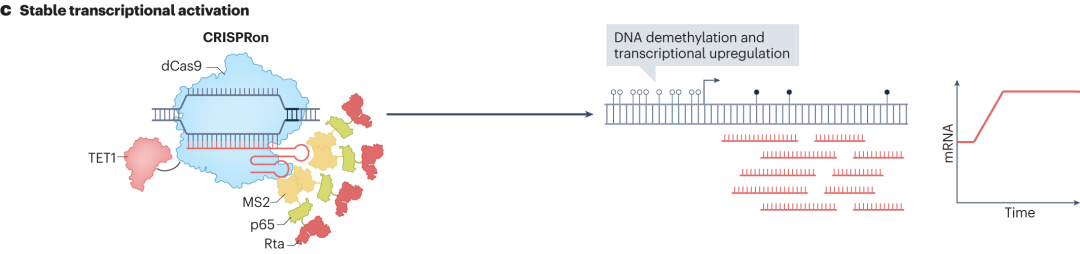
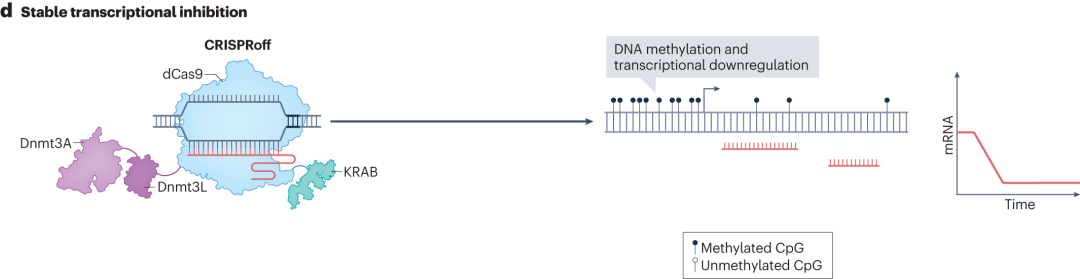
Although CRISPRa and CRISPRi allow programmable transcriptome engineering, they require continuous expression of dCas9 fusion proteins to maintain transcriptional control, which poses certain limitations in practical applications such as clinical therapies. To address this, researchers have developed CRISPRoff and CRISPRon technologies.
CRISPRon involves the fusion of optimized dCas9 with the catalytic domain of the TET1 DNA demethylase and the MS2 sgRNA scaffold.This catalyzes DNA demethylation, promotes the binding of transcription-related factors, and thus activates transcription. In addition, fusing dCas9 with hyperactive histone kinases (MSK1) or histone acetyltransferases (p300/CBP) can induce phosphorylation or acetylation of target histones, respectively. These histone modifications alter chromatin structure and function, making genes more accessible to transcription factors and thereby activating gene expression.
CRISPRoff consists of fusing the KRAB, Dnmt3A, and Dnmt3L protein domains with dCas9. Under the guidance of sgRNA, the complex binds to the target genomic region, where the Dnmt3A and Dnmt3L domains catalyze DNA methylation.Transient expression of CRISPRoff can establish stable DNA methylation at specific genomic regions, leading to transcriptional repression that is stably maintained during cell division and stem cell differentiation.
The discovery of RNA-targeting CRISPR nucleases has opened up a new frontier for transcriptome engineering. Cas13a, a single-effector nuclease derived from the Class II, Type VI CRISPR system, specifically recognizes and binds to target RNA sequences via programmable gRNAs. The nuclease domain of Cas13a then cleaves the target RNA, leading to downregulation of gene expression.However, after cleaving the target RNA, Cas13a exhibits collateral cleavage activity, which results in non-specific degradation of surrounding RNAs. This off-target activity may disrupt the physiological function of cells by degrading a large number of non-target RNAs.
Subsequently discovered homologs such as Cas13b and Cas13d demonstrate higher knockdown efficiency, yet they also suffer from collateral cleavage activity. To address this issue, researchers engineered variants of RfxCas13d with site-specific mutations in its nuclease domain, altering its spatial conformation and substrate-binding specificity to effectively reduce non-specific RNA binding and collateral activity. In addition, the discovery of the compact Cas13 nuclease Cas13X has provided new tools; structural and functional optimization of Cas13X has led to high RNA knockdown efficiency with greatly reduced collateral cleavage.
Furthermore, Cas7-11 is a novel RNA-targeting CRISPR nuclease from the Class I, Type III-E CRISPR system. It is capable of cleaving target RNAs without collateral activity, reducing potential cellular toxicity. Other type III effector complexes, such as CRISPR-Csm, can also be used for RNA knockdown and have shown good performance in zebrafish cells, E. coli, and mammalian cells, offering more options for RNA knockdown technologies across various biological models.
Figure 12. RNA Knockdown
2. RNA Base Editing
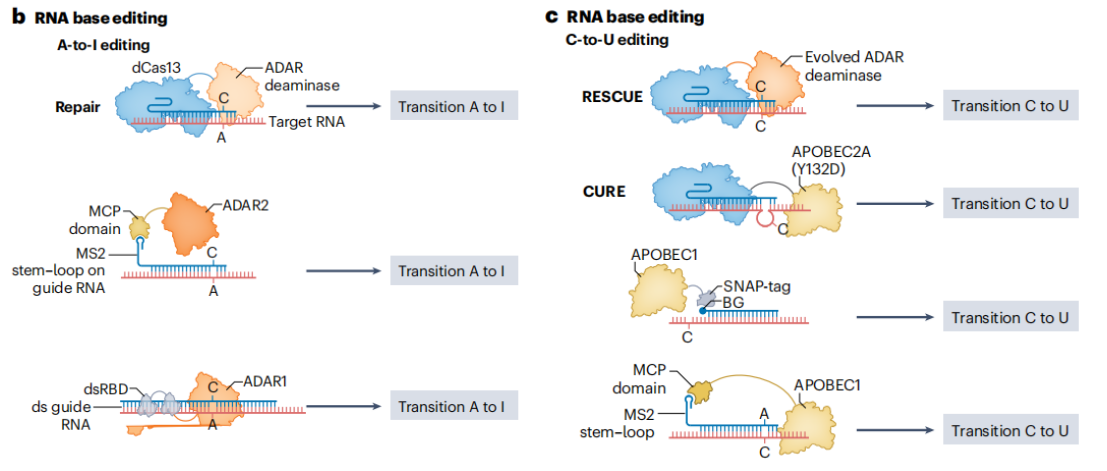
RNA editing tools based on the ADAR (Adenosine Deaminase Acting on RNA) family of deaminases enable A-to-I editing on double-stranded RNA (dsRNA) substrates.ADAR enzymes recognize adenosines within dsRNA and convert them to inosines, which are interpreted as guanosines during translation, thereby achieving base conversion at the RNA level. There are multiple strategies to achieve ADAR-based editing. One approach involves covalently linking ADAR to a gRNA or using high-affinity interactions via MS2 or BoxB domains to recruit endogenous ADAR enzymes to the target site. This method takes advantage of the cell’s own ADAR resources, reducing the need for exogenous proteins and minimizing potential immunogenicity and cytotoxicity. The REPAIR system fuses dCas13b with a hyperactive variant of ADAR2, enabling base editing at specific RNA sites to correct disease-associated mutations. However, the initial version of REPAIR suffered from off-target editing, as the hyperactive ADAR2 could also act on unintended sites, potentially disrupting normal gene expression. Rational protein engineering of ADAR2 led to the development of REPAIR v2, which significantly reduced off-target effects.
To achieve C-to-U editing, researchers evolved ADAR2 to possess cytidine deaminase activity and fused it with dCas13 to create the RESCUE system. By introducing specific mutations into ADAR2, RESCUE allows recognition and deamination of cytidines, converting them into uridines. To further reduce off-target effects, additional mutations were incorporated to lower the activity of ADAR2 at non-target sites. Moreover, by replacing dCas13 with the more compact EcCas6e protein, a highly efficient RNA base editor named ceRBE was developed. ceRBE exhibits excellent in vivo editing efficiency and reduced transcriptome-wide off-target effects.
Figure 13. RNA Base Editing
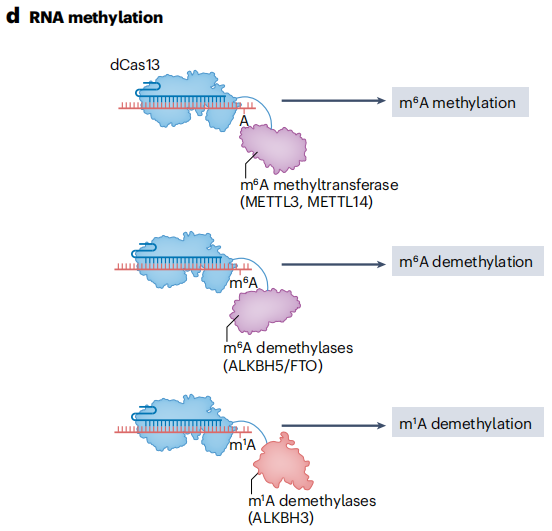
3. RNA Modification
Fusion of dCas13 with a truncated METTL3 methyltransferase domain enables the installation of N⁶-methyladenosine (m⁶A) modifications on specific RNA transcripts. m⁶A modifications play a critical role in regulating transcript abundance and alternative splicing, and they can affect RNA stability, translation efficiency, and subcellular localization. Through such fusion proteins, researchers can introduce m⁶A marks on specific RNA molecules to investigate their roles in gene expression regulation.
The REMOVER tool utilizes CRISPR-Cas13d to achieve targeted demethylation of N¹-methyladenosine (m¹A) sites, helping to elucidate the mechanisms by which m¹A influences RNA fate and gene expression. By fusing RNA demethylases such as Fat mass and obesity-associated protein (FTO) or AlkB homolog 5 (ALKBH5) with Cas13, researchers have achieved programmable, site-specific removal of m⁶A modifications. This provides a powerful tool for studying the dynamic regulation and functions of m⁶A. As RNA modification editors continue to be optimized, they are expected to play a greater role in transcriptome-wide phenotypic screening and in unraveling the functions of specific modifications in various cellular pathways.
Figure 14. RNA Methylation Modification
IV. Clinical Applications
With its programmability and powerful activity, CRISPR technology demonstrates tremendous potential in clinical treatment, encompassing key areas such as ex vivo cell engineering and in vivo gene correction.
In cancer immunotherapy, multiple clinical trials are currently underway. By using Cas9 nuclease ex vivo to knock out immunosuppressive genes in T cells, such as programmed cell death protein 1 (PD-1), the activity of T cells can be enhanced, enabling them to more effectively recognize and kill tumor cells. Additionally, knocking out the native T cell receptor (TCR) and introducing engineered TCRs or chimeric antigen receptors (CARs) allows T cells to specifically recognize antigens on the surface of tumor cells, significantly improving their targeting and cytotoxicity. Some patients have experienced clinical remission following such treatments.
In the treatment of monogenic blood disorders, CRISPR technology has also shown great promise. Sickle cell disease and transfusion-dependent β-thalassemia are associated with mutations in hemoglobin genes. By editing hematopoietic stem cells (HSCs) with CRISPR-Cas9 to disrupt regulatory elements of relevant genes, such as BCL11A, fetal hemoglobin expression can be upregulated as a compensatory mechanism, thereby alleviating anemia symptoms in patients.
For in vivo gene correction, CRISPR technology is primarily applied to treat genetic diseases in tissues that are relatively accessible for CRISPR nuclease delivery. For example, lipid nanoparticles (LNPs) can be used to deliver Cas9 mRNA and sgRNA to treat transthyretin amyloidosis by knocking out the transthyretin (TTR) gene in the liver, reducing toxic protein levels and alleviating organ damage. Similarly, LNP-mediated delivery of Cas9 to knock out the kallikrein gene can relieve symptoms of hereditary angioedema.
V. Conclusion
CRISPR technology has made significant progress in the fields of genome, epigenome, and transcriptome editing, leading to the development of various editing tools that have advanced both basic research and clinical treatments, showing great potential in areas such as cancer immunotherapy and the treatment of monogenic blood disorders. However, this technology faces challenges in clinical applications, including gene delivery, immunogenicity, editing accuracy, and safety. Future efforts should focus on optimizing editing tools by integrating machine learning and sequencing technologies, developing novel delivery vectors, exploring new gene targets, and combining them with cell-state response tools, while also addressing ethical regulation. This will help drive the broader application of CRISPR technology in medicine and biotechnology. References
Villiger L, Joung J, Koblan L, Weissman J, Abudayyeh OO, Gootenberg JS. CRISPR technologies for genome, epigenome and transcriptome editing. Nat Rev Mol Cell Biol. 2024 Jun;25(6):464-487. doi: 10.1038/s41580-023-00697-6. Epub 2024 Feb 2. Erratum in: Nat Rev Mol Cell Biol. 2024 Jun;25(6):510. doi: 10.1038/s41580-024-00745-9. PMID: 38308006.