- E-mail:BD@ebraincase.com
- Tel:+8618971215294
Epilepsy is a common neurological disorder affecting an estimated 65 million people worldwide. It is characterized by spontaneous and recurrent seizures, primarily driven by increased neuronal activity in the brain. Among the various types of epilepsy, temporal lobe epilepsy (TLE), which originates from one or more anatomical areas in the temporal lobe, is the most prevalent drug-resistant form of epilepsy in adults. In certain cases, removing the amygdala (amygdalectomy) alone is sufficient to eliminate seizures. The amygdaloid complex is structurally diverse, consisting of more than 10 nuclei; however, the specific nuclei and circuits responsible for seizures have not yet been identified.
On October 31, 2024, the team led by Xiaoming Li and Jiadong Chen from the College of Brain Science and Brain Medicine and the Center for Brain Research and Intelligence at Zhejiang University published a research paper in Advanced Science, titled “Posterior Basolateral Amygdala is a Critical Amygdaloid Area for Temporal Lobe Epilepsy.” This study reveals the neural circuit of glutamatergic neurons in the posterior basolateral amygdala (pBLA) in the occurrence and spread of epilepsy. Using a TLE mouse model, the study identifies pBLA as a critical nucleus within the amygdaloid complex that regulates seizures.

To manipulate glutamatergic neurons in the pBLA of C57BL/6J mice, researchers used an adeno-associated virus (AAV) tool mediated by the CaMKIIα promoter. AAV-CaMKIIα-ChR2-mCherry or AAV-CaMKIIα-mCherry was injected into the pBLA of the mice, and optical fibers were implanted bilaterally to stimulate the glutamatergic neurons in the pBLA via optogenetics (Figure 1A). Electroencephalography (EEG) recordings revealed that optogenetic activation of pBLA glutamatergic neurons induced significant epileptiform discharges lasting 39.75±5.31 seconds (n=6 mice) (Figure 1D). This activation significantly increased the power of brain waves across five different frequency bands: δ (0.5-4 Hz), θ (4-8 Hz), α (8-12 Hz), β (12-30 Hz), and γ (30-500 Hz) (Figure 1E). The activation also triggered grade 5-6 seizures (Figure 1F) and led to additional seizure symptoms, including pupil dilation and foaming at the mouth. Notably, a brief 30-second activation of pBLA glutamatergic neurons could result in sudden death (seizure stage 6; three out of six mice). In contrast, optogenetic activation of glutamatergic neurons in the anterior basolateral amygdala (aBLA) did not induce seizures.
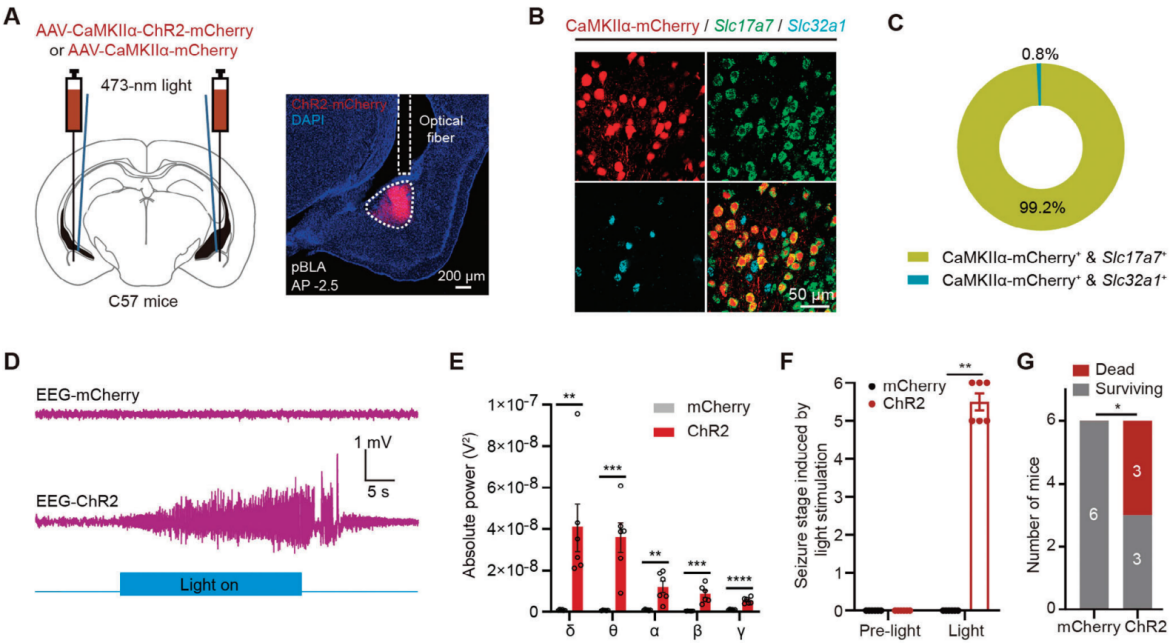
Figure 1. Optogenetic activation of glutamatergic neurons in the pBLA induces epileptic symptoms and even death
Epileptic seizures are characterized by sudden and brief episodes, with symptoms arising from excessive firing and synchronized neuronal activity in the brain. Therefore, glutamatergic neurons in the pBLA may send collateral projections to different brain regions, mediating this synchronous neuronal activity. To investigate potential collateral projection patterns, researchers injected a mixture of Retro-AAV-hSyn-Cre and AAV/9-DIO-EGFP into the BNST, and AAV-DIO-mCherry into the pBLA (Figure 2A). They found that pBLA→BNST neurons (pBLA neurons retrogradely labeled from the BNST) projected collaterals to the mPFC, IC, BNST, CeA, and VMH (Figure 2B). Additionally, double retrograde labeling experiments were conducted in the same mice, tracing back from different downstream targets: BNST and CeA, IC and BNST, as well as IC and CeA. These experiments revealed significant overlap among labeled pBLA neurons, with 69% of neurons double-labeled from BNST and CeA, 60% from BNST and IC, and 63% from IC and CeA.
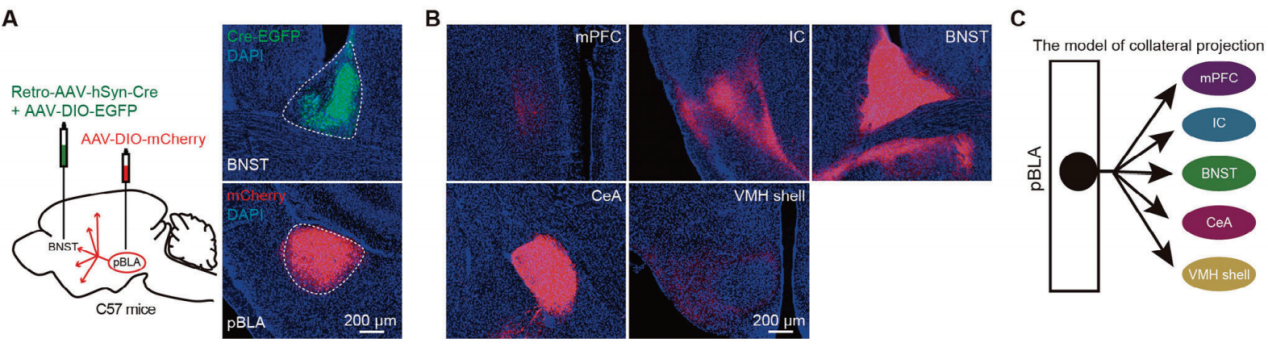
Figure 2. Retrograde labeling of pBLA projection neurons using AAV.
The researchers injected Retro-AAV-hSyn-Cre into the BNST and AAV-DIO-GCaMP6s into the pBLA to trace calcium activity in these neurons (Figure 3D). Using an in vivo fiber photometry recording system, they monitored calcium activity of collaterally projecting pBLA neurons in an acute epilepsy model induced by kainic acid (KA) injection in the hippocampus. By combining calcium signals with EEG recordings, they observed that the calcium activity of collaterally projecting pBLA neurons synchronized with EEG signals, showing a positive correlation (Figure 3E-G). In contrast, the calcium activity of aBLA→BNST neurons did not show strong correlation with EEG signals in the KA-induced acute epilepsy model (Figure 3H-K). Additionally, optogenetic activation of the aBLA-BNST circuit did not induce epileptiform discharges or seizures.
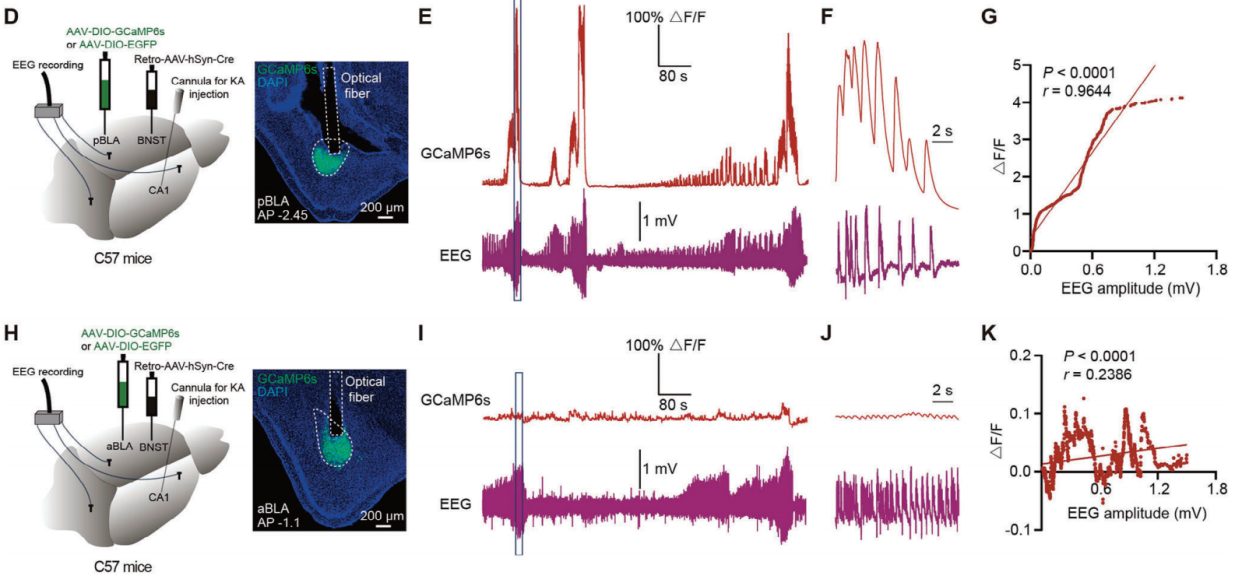
Figure 3. Calcium signals of collaterally projecting pBLA neurons synchronize with seizure activity.
Research demonstrated that optogenetic activation of pBLA glutamatergic axon terminals projecting to the IC, BNST, or CeA can induce epileptiform discharges and seizure behaviors. To further pinpoint which of these regions serves as the critical downstream area for seizure initiation by pBLA glutamatergic neurons, researchers selectively activated neurons within the IC, BNST, or CeA that receive projections from the pBLA. Antero-AAV2/1-hSyn-Cre was injected into the pBLA, and AAV-DIO-ChR2-EGFP was injected into the IC, BNST, or CeA, with optical fibers implanted above these regions (Figure 4A, B). Optogenetic activation of IC neurons targeted by the pBLA led to significant epileptiform discharges, seizure behaviors, and increased power across five EEG frequency bands (Figure 4C-E). In contrast, activation of BNST or CeA neurons receiving pBLA projections did not induce epileptiform discharges or seizures, nor did it significantly increase power in any EEG frequency band (Figure 4F-K). These findings suggest that the IC is the critical downstream region for seizure induction by pBLA glutamatergic neurons. Apoptosis of IC neurons reduced epileptiform discharges triggered by pBLA activation, significantly lowered power across the five EEG bands during seizures, and markedly decreased seizure severity, while apoptosis of BNST or CeA neurons did not produce these effects.
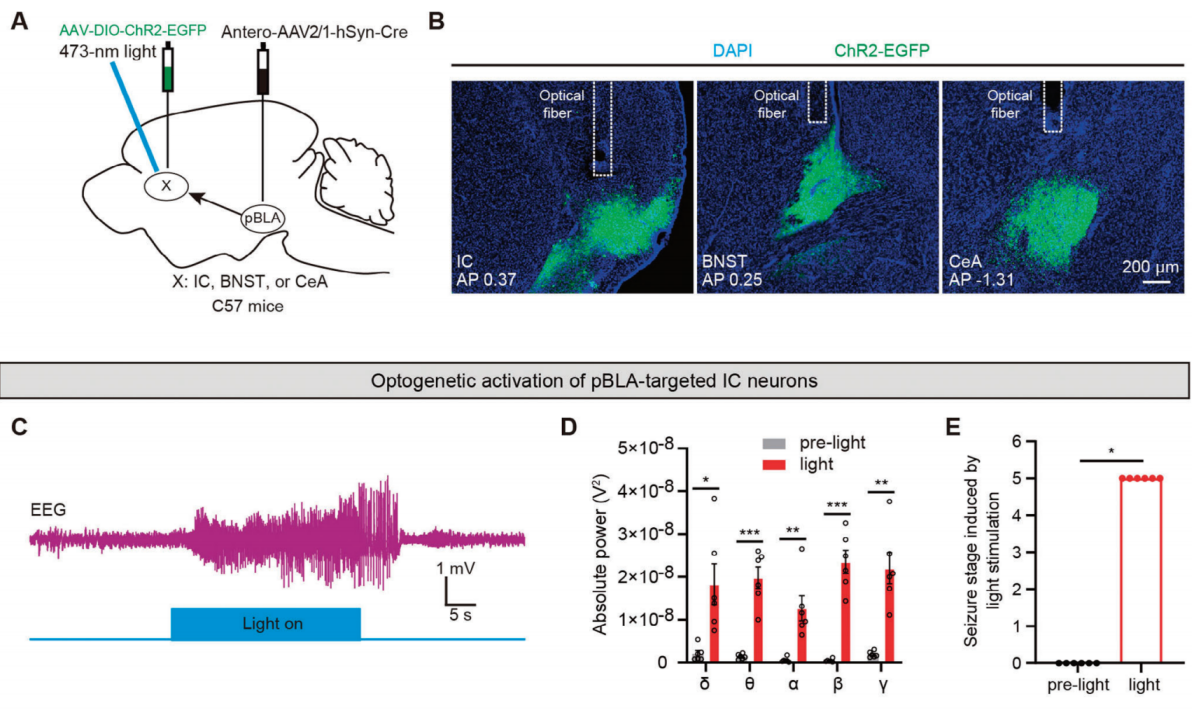
Figure 4. Optogenetic activation of pBLA neurons targeting the IC induces epileptiform discharges and seizures.
There are reciprocal projections between BNST GABA+ neurons and pBLA neurons, as well as between CeA GABA+ neurons and pBLA neurons. To determine which types of neurons in pBLA receive feedback inhibition from BNST GABA+ or CeA GABA+ neurons, the researchers injected a mixture of anterograde Antero-AAV-hSyn-EGFP and retrograde Retro-AAV-hSyn-mCherry into the BNST or CeA of C57BL/6J mice (Figure 5A, H). Neurons in pBLA that receive projections from BNST GABA+ or CeA GABA+ neurons were labeled with EGFP, while pBLA neurons that project to BNST GABA+ or CeA GABA+ neurons were labeled with mCherry (Figure 5B, I). FISH staining showed that BNST GABA+ or CeA GABA+ neurons formed synaptic connections with glutamatergic neurons in pBLA, and most of the pBLA glutamatergic neurons projecting to BNST GABA+ or CeA GABA+ neurons also received feedback projections from these GABAergic neurons.
To further determine the synaptic connection properties between BNST GABA+ or CeA GABA+ neurons and pBLA glutamatergic neurons, the researchers performed whole-cell patch-clamp electrophysiology experiments (Figure 5D, K). The results showed that optogenetic stimulation of axon terminals projecting from BNST or CeA to pBLA induced inhibitory postsynaptic currents (eIPSCs) in more than 70% of the mCherry-labeled pBLA glutamatergic neurons, rather than excitatory postsynaptic currents (eEPSCs) (Figure 5E, G, L, N). Moreover, optogenetic activation of pBLA glutamatergic neurons with projections to BNST GABA+ or CeA GABA+ axon terminals displayed monosynaptic inhibitory responses. The recorded eIPSCs were blocked by the voltage-gated sodium channel blocker tetrodotoxin (TTX, 1μM), then restored by the potassium channel blocker 4-aminopyridine (4-AP, 100μM), and abolished by the GABAA receptor antagonist picrotoxin (PTX, 100μM) (Figure 5E, F, L, M). Combining viral tracing, FISH, and patch-clamp electrophysiological results, it was concluded that BNST GABA+ or CeA GABA+ neurons form feedback inhibition to pBLA glutamatergic neurons (Figure 5B, C, I, J).
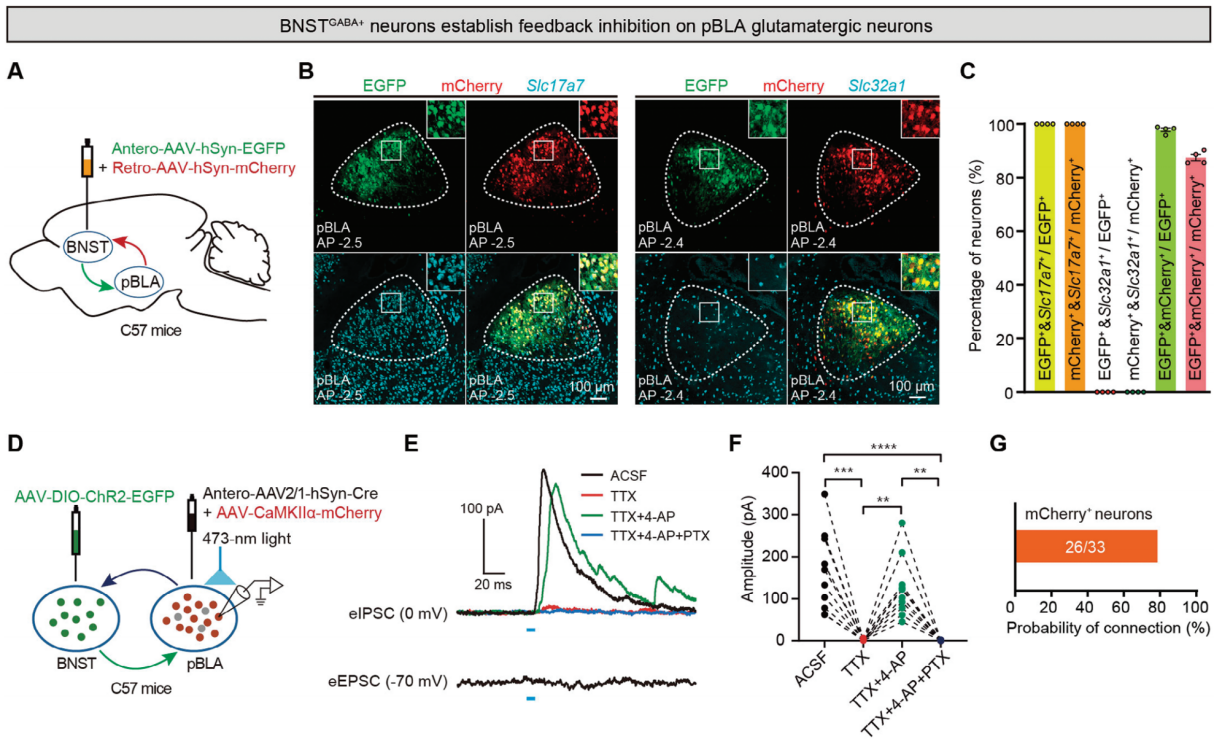
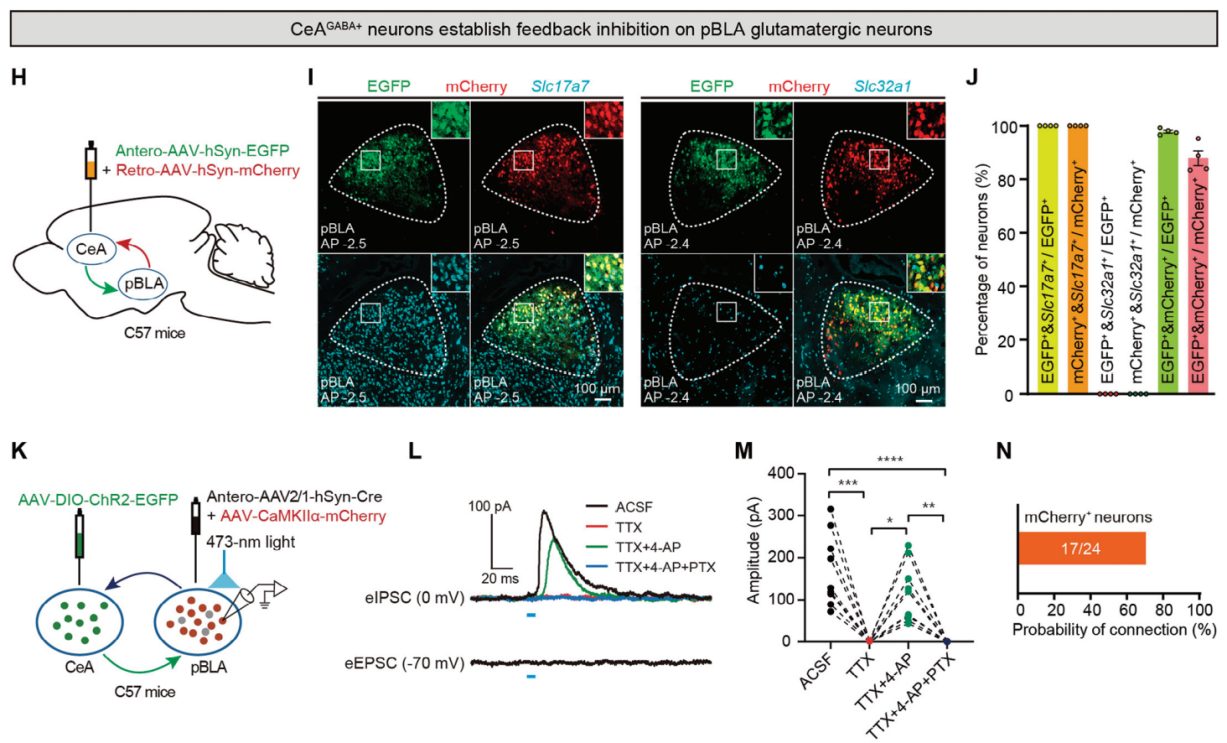
Figure 5. Feedback inhibition of pBLA glutamatergic neurons by BNST GABA+ and CeA GABA+ neurons.
This figure demonstrates the use of rabies virus (RV) retrograde monosynaptic tracing to identify upstream inputs to pBLA glutamatergic neurons. Retro-AAV-hSyn-Cre was injected into BNST, and Cre-dependent RV helper viruses were injected into pBLA, followed by the injection of G-protein-deficient rabies virus (RV-EnvA-ΔG-dsRed) into pBLA. The results show that pBLA glutamatergic neurons receive excitatory input from the ventral hippocampus CA1 (vCA1) region. Furthermore, anterograde tracing with AAV-CaMKIIα-mCherry into vHIP confirmed widespread excitatory projections from vCA1 to pBLA but not to other amygdala regions like aBLA and CeA.
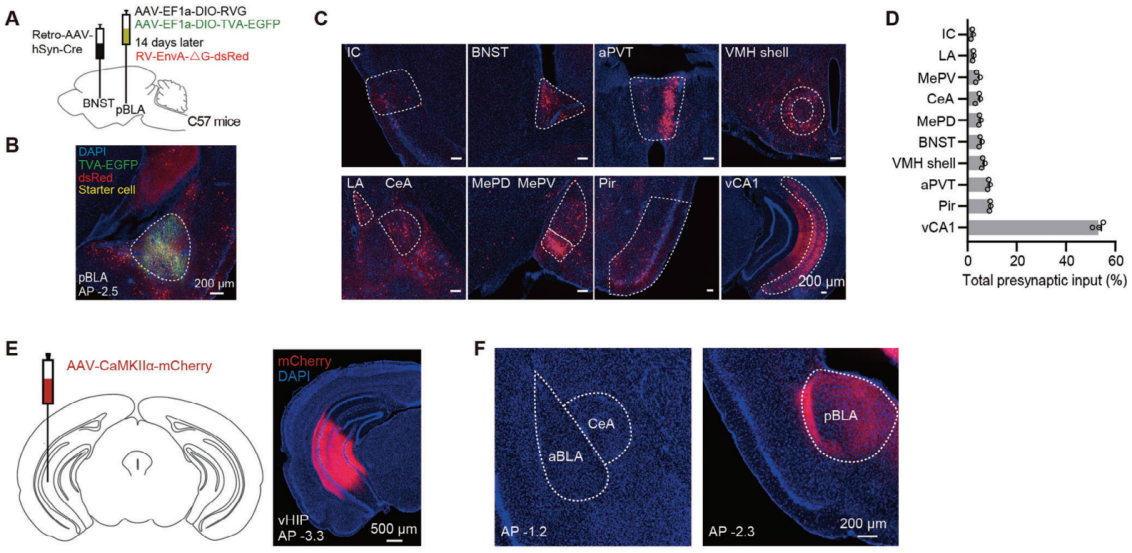
Figure 6. Monosynaptic retrograde tracing of projections from pBLA neurons to BNST.
To test if activation of the vCA1-pBLA circuit is sufficient to trigger seizures, the researchers injected AAV-CaMKIIα-ChR2-mCherry or AAV-CaMKIIα-mCherry into the vCA1 of C57BL/6J mice and implanted optical fibers above the pBLA. Optogenetic activation of the vCA1-pBLA circuit induced epileptiform discharges and seizure-like behaviors. In contrast, although pBLA neurons also receive synaptic inputs from the piriform cortex (Pir) and anterior paraventricular thalamic nucleus (aPVT) (Figure 7G), optogenetic activation of the excitatory Pir-pBLA or aPVT-pBLA circuits did not induce epileptiform discharges or seizures (Figures 7H-J).
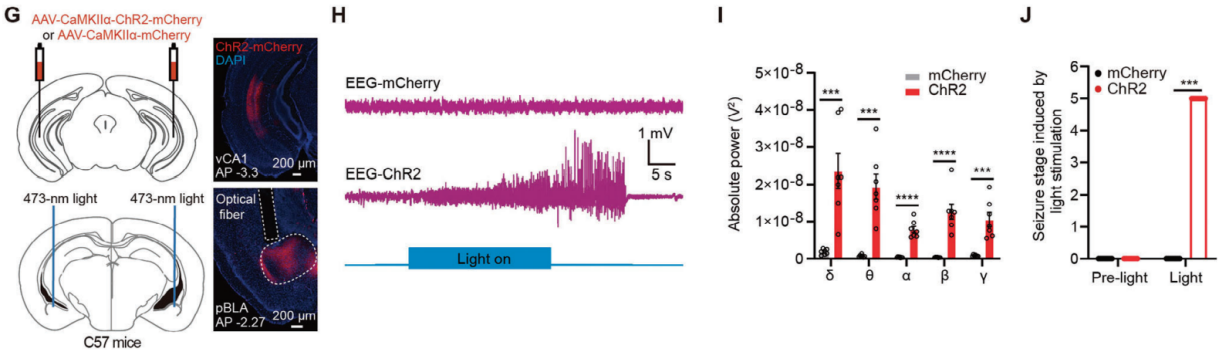
Figure 7. Optogenetic activation of the vCA1-pBLA circuit.
To investigate whether the ablation of glutamatergic neurons in the pBLA can reduce seizures in a temporal lobe epilepsy (TLE) mouse model induced by KA injection into the hippocampus, the researchers injected a mixture of AAV-DIO-DTA and AAV-CaMKIIα-Cre into both sides of the pBLA (Figure 8B). Four weeks after viral injection, a reduction in NeuN-positive cells in the pBLA region was observed in the mice injected with DTA. After KA was administered to the hippocampal CA1 region through the implanted catheter, the mice were monitored for 60 minutes to assess epileptic behavior and EEG signals on the skull surface (Figure 8A). Mice with ablation of pBLA neurons showed significant reductions in EEG power spectra and seizure severity (Figures 8C, D). Ablation of pBLA neurons also significantly reduced fatal outcomes during acute seizures (Figure 8E). Frequency spectrum analysis of the EEG signals indicated a substantial decrease in the power of all five types of brain waves (Figure 8F). Overall, the ablation of pBLA glutamatergic neurons is a potent intervention that can suppress KA-induced acute seizures and reduce the risk of severe epilepsy-associated death.
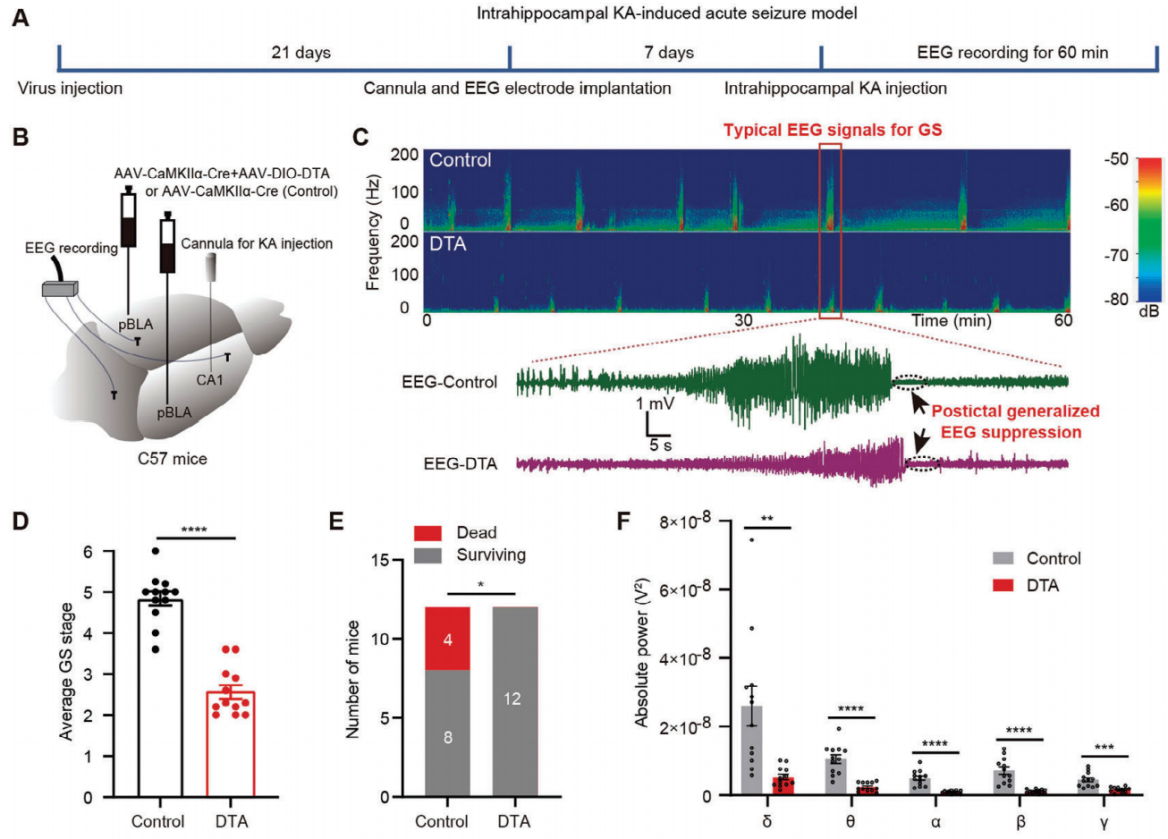
Figure 8. Ablation of pBLA glutamatergic neurons suppresses KA-induced acute seizures.
Next, the researchers explored whether ablation of pBLA glutamatergic neurons could suppress KA-induced chronic spontaneous seizures. KA was injected into the hippocampus, and AAV-DIO-DTA and AAV-CaMKIIα-Cre were bilaterally introduced for pBLA neuron ablation. Electrodes were placed on the skull surface for EEG recording. Seven weeks after KA injection, the mice were assessed daily for more than 8 hours, with spontaneous high-voltage spikes (> 0.5 mV) evaluated for 5 consecutive days (Figure 9G). Ablation of pBLA glutamatergic neurons significantly reduced the number of spontaneous high-voltage spikes (Figure 9H, I). No GSs were observed in the pBLA ablated mice (Figure 9J, K). These findings suggest that ablation of pBLA neurons effectively alleviates seizures in the chronic TLE model.
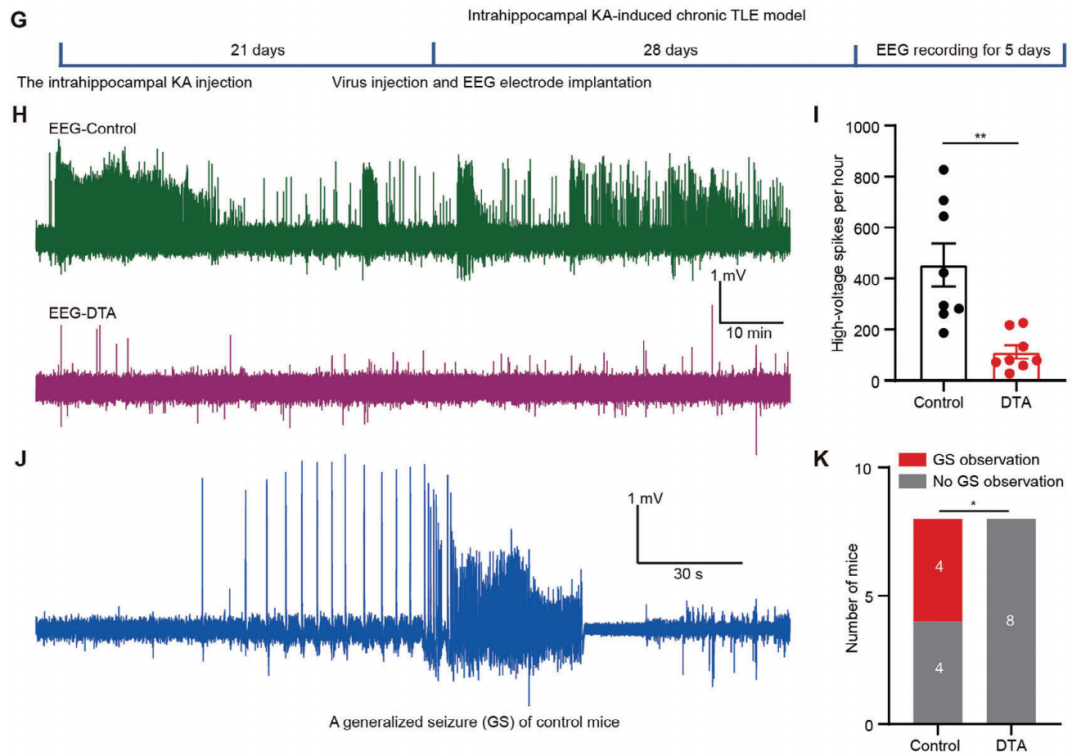
Figure 9. Ablation of pBLA neurons alleviates seizures in the chronic TLE model.
For more details, please refer to the original text.
Brain Case can provide and customize related products. If needed, you can consult BD@ebraincase.com

WhatsApp Business Account
Address:-
Address:-



